 Natalia S Araujo wants to find out, and she’s not the only one. As the only bee species in which true polygyny (multiple fertile queens in the same colony) occurs, there is great interest in Melipona bicolor, and its mitochondrial genome (mt genome) was one of the first sequenced in bees. But the sequence was incomplete and lacked information about its mitochondrial gene expression pattern.
Natalia S Araujo wants to find out, and she’s not the only one. As the only bee species in which true polygyny (multiple fertile queens in the same colony) occurs, there is great interest in Melipona bicolor, and its mitochondrial genome (mt genome) was one of the first sequenced in bees. But the sequence was incomplete and lacked information about its mitochondrial gene expression pattern.So Araujo, a postdoctoral researcher of animal genomics in the GIGA Institute of the University of Liège, Belgium, and her collaborator, Maria Cristina Arias from the University of Sao Paulo, Brazil, combined long and short reads of M. bicolor DNA with RNASeq data to characterize its species control region and transcription patterns.
Araujo reported the results at the fourth annual SMRT Leiden Scientific Symposium, held May 7-8 at Leiden University Medical Center in the Netherlands, the first day of which was abuzz with research that highlighted the importance of biodiversity and conservation genomics.
They created a 15,001bp mt genome, including a control region of 255 bp, with the highest AT content reported so far for bees (98.1%).
“Interestingly, conserved structures were identified for the first time in the control region of all eusocial corbiculate bees sequenced so far,” Araujo said. “M. bicolor has one of the most compact and functional mtDNA reported in bees. Results reveal unique and shared features of the mitochondrial genome in terms of sequence evolution and gene expression making M. bicolor an interesting model to study mitochondrial genomic evolution.”
Illuminating Life with Better Sequencing
SMRT Leiden Keynote speakers Paul Hebert of the University of Guelph, Canada, and Mara Lawniczak of the Wellcome Sanger Institute, UK, bookended the day with inspiring talks about large-scale species sequencing projects.
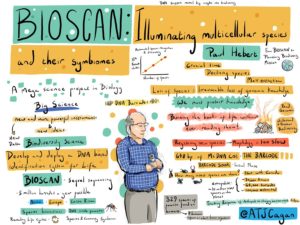 Hebert introduced BIOSCAN, an initiative by the International Barcode of Life (iBOL) project to use new technology to identify, register, and monitor life. Launched in 2007 out of Leiden, the iBol consortium, had the original mission to develop and deploy a DNA-based “barcode” identification system for animal, fungi, and plants. It successfully delivered the DNA barcode for 500,000 species in 2015, and developed an automated species recognition & discovery system called Barcode of Life Data System (BOLD), which will now help them focus on scanning for the unknown.
Hebert introduced BIOSCAN, an initiative by the International Barcode of Life (iBOL) project to use new technology to identify, register, and monitor life. Launched in 2007 out of Leiden, the iBol consortium, had the original mission to develop and deploy a DNA-based “barcode” identification system for animal, fungi, and plants. It successfully delivered the DNA barcode for 500,000 species in 2015, and developed an automated species recognition & discovery system called Barcode of Life Data System (BOLD), which will now help them focus on scanning for the unknown.
Originally based on Sanger sequencing, iBOL moved to using the PacBio Sequel System for species discovery after finding that it was more cost-effective and accurate at delivering species-level information. They now use it to barcode 5 million species per year and run species discovery for 500,000 species per year.
“There’s a real urgency in this that I haven’t encountered in other areas of science, in that the subjects of our research are disappearing. Don’t let the dodo fool you – most animals that go extinct don’t leave their bones behind on the forest floor. They leave a smudge,” Hebert said. “Every loss of species is a valuable loss of genomic data.”
They will also be using the Sequel System to elucidate species interaction. Environmental samples are messy, Hebert told the SMRT Leiden crowd. Some of the insect samples his team analyzed had DNA mixtures from animals they fed on (elephants, kangaroo, even nematodes). So, to characterize comprehensively, they extract the DNA of whole species, amplify with primers from different taxa, sequence deeply (>3,000-fold), and create species symbiomes.

Lawniczak discussed the Earth BioGenome Project, a moonshot for biology aiming to sequence, catalog, and characterize the genomes of all of Earth’s eukaryotic species, and Sanger’s own Darwin Tree of Life Project.
She also went into detail about efforts to better understand disease vector species through genetic sequencing, including the Anopheles gambiae 1000 Genome Project and the Sanger project to sequence the A. coluzzi genome from a single mosquito using PacBio’s new low DNA input workflow.
“How can we outpace the evolution of resistance?” she asked the crowd. The answer: high-quality reference genomes and de novo insect assemblies.
Other speakers described how long-read technology is revealing new insights into the diverse species that are relevant to our nutrition (bitter gourd by Henri Van de Geest of Genetwister Technologies, Netherlands, and cassava by Herve Vanderschuren of the University of Leige, Belgium), medicine (cannabis by Kevin McKernan of Medicinal Genomics), and evolution (cave fish by former SMRT Grant winner Fritz Sedlazeck of the Baylor College of Medicine Human Genome Sequencing Center).
Presentations by Kateryna Makova of Penn State, Oliver Duss of the Scripps Research Institute, and Iain Macaulay of the UK’s Earlham Institute, also revealed how SMRT Sequencing can be developed to query chemical processes (transcription) and reveal new information (single cell).
For an in-depth report on each presentation from Day One, check out this blog post by PacBio Principal Scientist Elizabeth Tseng.
Evolving Towards Precision Medicine
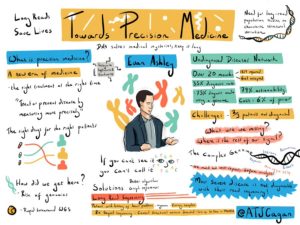 If there’s one area where accuracy is essential, it’s medicine, as highlighted by Euan Ashley as he opened Day Two of SMRT Leiden.
If there’s one area where accuracy is essential, it’s medicine, as highlighted by Euan Ashley as he opened Day Two of SMRT Leiden.
“If you miss one important coding variant in one gene, it could be the difference between life and death,” the cardiologist said.
A bit of a DNA detective, Ashley described his work at Stanford and as part of the Undiagnosed Diseases Network using long-read sequencing to help solve medical mysteries. Within a 20-month period, they delivered diagnoses for 132 out of 382 cases, a 35% solve rate, and a surprising 79% of the solved cases had actionability.
“This is truly transformational technology,” Ashley said. “There has been a lot of hype around genomics, but this is actual delivery here.”
What are we missing in the other 65% of cases? Repeat sequences, paralogous genes, mosaicism, non-disruptive variants — complexities of the genome that require long, comprehensive reads, better algorithms, and, ideally, graph reference genomes, he said.
 Evan Eichler of the University of Washington, an early adopter of PacBio technology, also discussed the challenges of characterizing structural variation (SV) in the human genome, and some of his solutions.
Evan Eichler of the University of Washington, an early adopter of PacBio technology, also discussed the challenges of characterizing structural variation (SV) in the human genome, and some of his solutions.
His lab’s first study using PacBio for SV calling was eye-opening because it found ~22,000 novel genetic variants corresponding to 11 Mb of sequence. They had not expected to see so much novelty.
However, even with the long reads, segmental duplications (SD) remain challenging. These SD regions encompass nearly 500 genes and are the most copy number polymorphic regions , so they are important to catch, yet Eichler found that 75% of SDs on a human genome he analyzed were not assembled. So he used a method called Segmental Duplication Assembly (SDA), which maps reads to assembled contigs to identify variants which can then be used to separate reads into paralogous sequences.
Other speakers showcased the use of long reads to resolve complex regions in the human genome that have both important evolutionary and clinical significance. Kornelia Leveling of Radboud University Medical Centre, discussed long-read amplicon sequencing and its advantages in clinical applications (less PCR, improved breakpoint detection and haplotyping, and the ability to separate paralogous genes) and Melissa Laird Smith of the Icahn Institute at Mt. Sinai, spoke about how she is using PacBio sequencing to characterize the full diversity of the immunoglobin heavy chain locus.
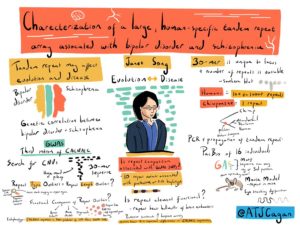 The SMRT Leiden audience also heard how accurate, long reads are being adopted for testing medically important genes that can have therapeutic actionability. Ming-Hsiang Lee of the Sanford Burnham Prebys Medical Discovery Institute presented work on somatic mosaicism in the APP gene in relation to sporadic Alzheimer’s disease. Janet Song of Stanford explained how she used long-read sequencing to study tandem repeats in the CACNA1C intronic region to better understand its association with bipolar disorder and schizophrenia.
The SMRT Leiden audience also heard how accurate, long reads are being adopted for testing medically important genes that can have therapeutic actionability. Ming-Hsiang Lee of the Sanford Burnham Prebys Medical Discovery Institute presented work on somatic mosaicism in the APP gene in relation to sporadic Alzheimer’s disease. Janet Song of Stanford explained how she used long-read sequencing to study tandem repeats in the CACNA1C intronic region to better understand its association with bipolar disorder and schizophrenia.
Alexander Mellmann of University Hospital Munster, spoke about tracking bacterial infections in hospitals by combining whole genome sequencing with genome-wide gene-by-gene typing (cgMLST). The pipeline has enabled the hospital to get better information about transmission rates and to evaluate its hygiene control methods.
And Yahya Anvar of the Leiden University Medical Center reported the use of PacBio HiFi reads for resolving complex pharmacogenes. Differential drug response is common (in 50% of the population) and dangerous (fourth leading cause of death). Previously, many of the PGx genes could not be characterized by short-read sequencing due to the genes’ highly repetitive nature, but Anvar showed that 77% of the important PGx genes could be fully phased, while another 19% could be partially resolved, using HiFi reads at 30-fold coverage.
For an in-depth report on each presentation from Day Two, check out this blog post by PacBio Principal Scientist Elizabeth Tseng.
Want a visual summary of the two-day conference? Evolutionary biologist and scientific illustrator Alex Cagan of the Sanger Institute was on site sketching each presentation in real-time – how appropriate. You can check out the resulting work here.
SMRTLeiden 2019 – Day 1 – Curated tweets by PacBio SMRTLeiden 2019 – Day 2 – Curated tweets by PacBio