Complete microbial genomes with ease and confidence
For most microbes, closed genomes with accessory plasmids can be assembled with one touch using the default settings of our assembly pipeline.
- Generate platinum-standard, closed reference genomes
- Identify ever-evolving genes associated with toxicity, virulence, and antimicrobial resistance
- Resolve strains, serotypes, and plasmids to track pathogen outbreaks in humans, plants, and animals, through food systems, hospitals, and communities
- Comprehensively characterize microbes to facilitate scientific breakthroughs
Workflow: From DNA to characterized microbial genomes in a single experiment

Library preparation
Starting with unamplified genomic DNA, prepare libraries for whole genome sequencing using standardized protocols and workflow recommendations.
- Prepare SMRTbell libraries with barcoded adapters following the protocol for microbial whole genome sequencing, or use the seqWell LongPlex multiplexing kit.

Sequencing
The Revio and Vega systems provide affordable high-quality genome assemblies.
- Simultaneously generate whole genome and epigenome data by sequencing on a PacBio HiFi long-read system
Data analysis
Assemble genomes with full-solution analytical software tools and standard file formats.
- Use SMRT Link for fully automated demultiplexing, assembly, circularization, and polishing of both chromosomes and plasmids to produce gold standard references
- Download and explore HiFi data from high-throughput bacterial whole genome sequencing
Webinar
MORE SAMPLES, LOWER COSTS, LESS TIME: NEW PACBIO HIFI PREP KITS + MICROBIAL WGS AND ANTI-MICROBIAL RESISTANCE
Learn how the HiFi plex prep kit 96 allows users to sequence small genomes at similar costs to short-read prep workflows, all while making high-quality sequencing more accessible by eliminating common bottlenecks related to cost, time, and labor.

Figure 6: A) Graph representation of the duplication. Nodes are numbered and their length in bp is indicated. The colors indicate sequence similarity as determined through a blast search of the duplicated and adjacent parts. B) A model of homologous recombination underyling the duplication.
Spotlight
VIRTUALLY BASE-PERFECT MYCOBACTERIUM TUBERCULOSIS COMPLEX (MTBC) GENOMES CAN BE ASSEMBLED FROM PACBIO HIFI READS
The accuracy of HiFi reads allowed researchers to forego many of the methodological complexities of previous approaches to long-read assembly. Noisy long reads necessitate high sequencing depths and hybrid approaches that combine long and short reads and multiple assembly and polishing steps. This results in complicated pipelines and directs attention away from biological to technical questions. They found that a depth of 16x with HiFi reads was sufficient to obtain a closed bacterial genome.
Stritt, C., et. al. (2025) Gene conversion and duplication contribute to genetic variation in an outbreak of Mycobacterium tuberculosis. Microbial genomics.
Spotlight
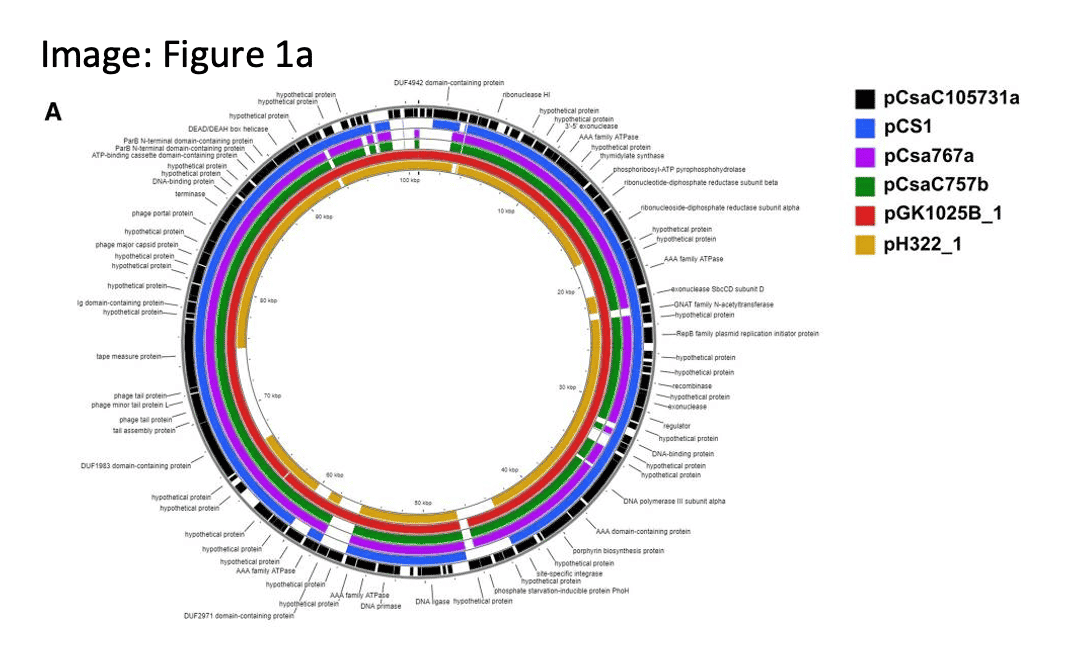
INVESTING IN MANUFACTURED FOOD SAFETY WITH NEW REFERENCE GENOMES
Cronobactoer sakazakii infections can cause lifelong consequences in pre-term or low birth-weight infants. Complete genomes and associated plasmids from 2 highly persistent C. sakazakii strains recovered from infant formula manufacturing facilities, encoding wide array of pathogenicity and virulence genes, will add to our understanding of pathogen adaptation and persistence in built environments.
Negrete, F.J. (2022). Complete genome sequences and genomic characterization of five plasmids harbored by environmentally persistent Cronobacter sakazakii strains ST83 H322 and ST64 GK1025B obtained from powdered infant formula manufacturing facilities. Gut Pathog.


Spotlight
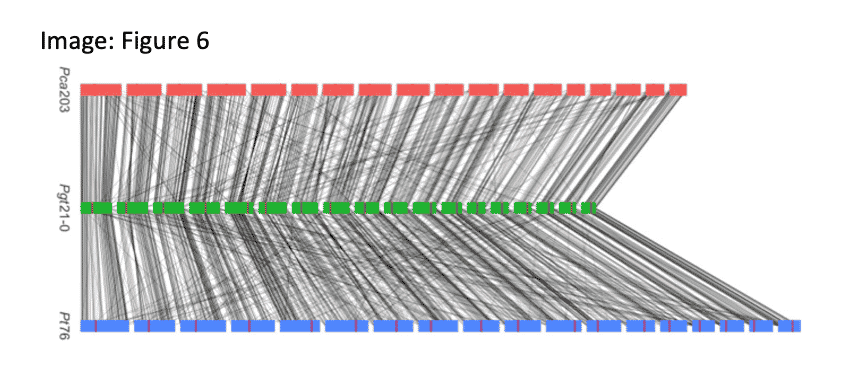
FULLY PHASED FUNGAL GENOMES FUEL INSIGHTS INTO VIRULENCE
A fully phased, 18-chromosome reference for the oat crown rust fungal pathogen Paccinia coronate f. sp. avenae (Pca203) shares high homology with the two other cereal rust fungi for which phased genomes are available. With 2,029 predicted effector genes, the new genome will aid in the discovery of alleles linked to growing virulence.
Henningsen, E.C. (2022). A chromosome-level, fully phased genome assembly of the oat crown rust fungus Puccinia coronata f. sp. avenae: a resource to enable comparative genomics in the cereal rusts. G3 journal.

{kind=link}
{kind=link}