UPDATED
August 12, 2019
This paper is now published in Nature Biotechnology.
ORIGINAL POST
January 15, 2019
The preprint describes a collaborative effort to comprehensively characterize a human genome — we chose the well-analyzed HG002/NA24385 sample available as a benchmark from the Genome in a Bottle consortium — Lead authors Aaron Wenger and Paul Peluso, senior authors David Rank and Michael Hunkapiller, and co-authors at PacBio, Google, NIST, and a host of leading academic institutions and companies contributed to the publication.
The work stems from our ongoing commitment to keep increasing the quality and usability of data generated from SMRT Sequencing systems. “Today, human genomes are sequenced at population scales, but it remains necessary to combine sequencing technologies to cover all types of genetic variation, which increases cost and adds complexity to projects,” the paper’s authors explain. “A sequencing technology with long read length and high accuracy would enable a single experiment for comprehensive variant discovery.”
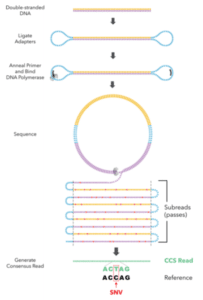
To that end, the team developed a new protocol based on the CCS method, which builds a consensus sequence based on many passes across the same template. “Recent gains in read length for SMRT Sequencing and optimized DNA template preparation suggested an opportunity to unify high accuracy with long read lengths using CCS,” the scientists report.
Using the human genome as a proving ground, the authors selected a library tightly-distributed at 15 kb, generated CCS reads with an average of 10 passes, and sequenced the genome to 28-fold coverage. The average read accuracy is 99.8%, matching the accuracy of the typical short read. De novo assembly of the reads yielded “a highly contiguous and accurate genome with contig N50 above 15 Mb and concordance of Q48 (99.998%),” they add.
The team also interrogated a broad range of variants and performed phasing. “We analyze the CCS reads to call SNVs, indels, and structural variants; to phase variants into haplotype blocks; and to de novo assemble the HG002 genome,” the scientists report. “The CCS performance for SNV and indel calling rivals that of the commonly-used pairing of BWA and GATK on 30-fold short-read coverage.” Detection of variants was consistently strong for SNVs (99.91%), indels (95.98%), and structural variants (95.99%). As the authors note, “Nearly all (99.6%) variants are phased into haplotypes, which further improves variant detection.”
Beyond the remarkable quality results from this protocol, the scientists note a number of other advantages with this approach. These include easier sample prep, since there is no need for ultra-long genomic DNA, reduced computational time, and the ability to use familiar tools like GATK designed for accurate reads.
Future improvements to the method — such as faster generation of HiFi reads from subreads and increasing the number of reads produced in a run [Update 8/12/19: The Sequel II System provides ~8 times more HiFi reads per run than the Sequel System used in this study] — should “facilitate rapid, population-scale analysis of full genomes to improve human health,” the authors write. The HiFi protocol also will have application outside of human genomics, with utility in metagenomics as well as plant and animal genome assembly.
January 15, 2019 | Human genetics research