 We were delighted to host an educational workshop at last month’s annual meeting of the American Society of Human Genetics (ASHG), where we had the opportunity to feature talks from two customers as well as an overview of SMRT Sequencing. If you couldn’t attend, check out the videos or read the highlights below.
We were delighted to host an educational workshop at last month’s annual meeting of the American Society of Human Genetics (ASHG), where we had the opportunity to feature talks from two customers as well as an overview of SMRT Sequencing. If you couldn’t attend, check out the videos or read the highlights below.
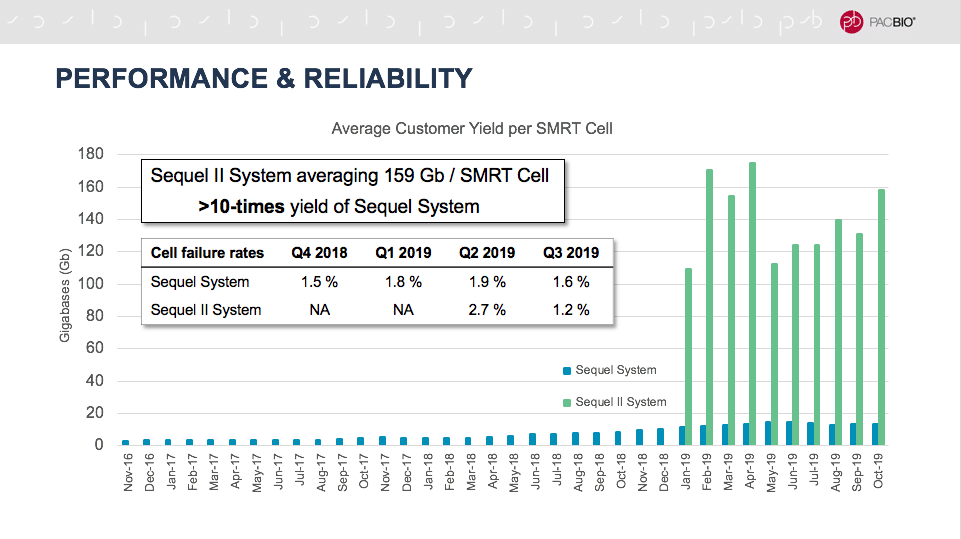
Emily Hatas, our director of business development, kicked things off with a look at how SMRT Sequencing has evolved over the years. Compared to the first instrument we offered, the Sequel II System represents a 100-fold improvement in read length and a 10,000-fold improvement in throughput. As of last month, customers were averaging about 160 Gb per SMRT Cell, a yield more than 10 times higher than the Sequel System.
Most of the presentation focused on applications in human genome analysis. High-throughput structural variant detection, which makes use of continuous long-read (CLR) sequencing, is well-suited to population studies and can be run at a cost of about $670 per sample when running two samples on each SMRT Cell. Comprehensive variant detection, which uses HiFi sequencing to make multiple passes around each molecule for optimal accuracy, is great for disease research — particularly for solving rare diseases — and costs about $2,600 per sample, assuming each library uses two SMRT Cells. Finally, de novo assembly of reference genomes should also be based on HiFi reads, Hatas told attendees, since it achieves comparable contiguity to CLR mode with about six times higher accuracy. In addition, HiFi data cuts analysis time in half and generate much smaller files to make de novo assembly more scalable.
Next up was Naomichi Matsumoto from Yokohama City University to speak about the use of SMRT Sequencing to solve Mendelian diseases. He shared the story of how his lab discovered a 12.4 kb structural variant that’s responsible for progressive myoclonic epilepsy in two siblings. The variant was in a repetitive, GC-rich region, which was why previous attempts to find it had failed. With low-coverage whole genome sequencing on the Sequel System, his team identified the variant and later confirmed that it was causal.
Matsumoto also reported progress in understanding repeat expansion disorders — many of which have neurological components — by pairing SMRT Sequencing with new analysis tools designed to highlight repetitive areas. In one example, his team was able to distinguish between the smaller number of repeats associated with healthy controls and the larger numbers associated with symptomatic patients.
The final talk came from Shawn Levy of the HudsonAlpha Institute for Biotechnology and the recently spun out services lab, now known as HudsonAlpha Discovery, which is a division of Discovery Life Sciences. He offered a look at his team’s early access experience with the Sequel II System, which was so successful that the research institute now has four of the instruments.
His data showed the increasing output of the system over time, as well as yield increases from the HiFi method. Levy noted that accuracy improves with each pass around the molecule, but reaches a plateau at the tenth pass or so. For Iso-Seq experiments, the team saw a significant improvement in yield from the Sequel System to the Sequel II System. Levy also shared hot-off-the-presses data from a project designed to determine the quality of Iso-Seq reads that can be gleaned from FFPE samples. The longer reads made possible with this approach don’t overcome the highly fragmented DNA and RNA coming from the samples, Levy said, but they definitely improve biological resolution and enable the characterization of higher molecular weight RNA that’s present in the samples. The project required a modified Iso-Seq protocol, which is still being optimized for best performance. While conventional approaches are evaluated based on how many 200-nucleotide reads they generate, the SMRT Sequencing method resulted in an average length of 435 bases.
Levy noted that his team also uses long-read sequencing for targeted sequencing applications associated with confoundingly homologous regions and for analyzing complex rearrangements in cancer. Going forward, they will also be sequencing about 7,000 genomes using long-read WGS for the All of Us Research Program to increase discovery of structural variants.
We’d like to thank all of the ASHG attendees who made our workshop such a success! If your research includes human genetics, please consider applying for our 2019 Human Genetics SMRT Grant Program. The winner will receive complimentary sequencing from the HudsonAlpha Genome Sequencing Center of up to 12 SMRT Cells. The deadline to apply is November 22, 2019.
November 12, 2019 | Events + conferences