Model organisms such as yeast, Arabidopsis and Drosophila have been essential to progress in genetic and biomedical research for more than 100 years. Model organisms are the best, fastest, most effective way to advance science especially when human experimentation may not be feasible. Numerous biological principles have been elucidated using model organisms, including Nobel-prize winning discoveries by Thomas Hunt Morgan that genes are carried on chromosomes; by Hermann Muller for the discovery that X-ray irradiation causes mutations; and by Edward B. Lewis, Christiane Nüsslein-Volhard, and Eric Wieschaus for their discoveries revealing the genetic control of early embryonic development – all using the fruit fly Drosophila melanogaster. D. melanogaster also played a crucial role in the development of genomics by being one of the first multicellular organisms to be sequenced and assembled by a whole genome shotgun method, followed by directed clone-based finishing. The resulting D. melanogaster reference genome sequence provides an invaluable test bed for developing new genome sequencing and assembly technologies.
In collaboration with Dr. Casey Bergman at the University of Manchester and Drs. Susan Celniker and Roger Hoskins of the Berkeley Drosophila Genome Project (BDGP) at Lawrence Berkeley National Laboratory, we have sequenced adult males from a subline of the ISO1 (y; cn, bw, sp) strain of D. melanogaster. This is the same stock used in the official BDGP reference assemblies since the first genome sequence release in 2000. The DNA was size-selected for >15 kb elution using the BluePippinTM system (Sage Science), and in total, ~15 Gb of sequence was generated from a 20 kb library using P5-C3 sequencing chemistry on the PacBio® RS II.
Total number of bases: 15,208,567,933 bp
Total number of reads: 1,514,730
Average read length: 10,040 bp
Half of sequenced bases in reads greater than: 14,214 bp
PacBio RS II instrument time for sequencing: 6 days
Number of SMRT® Cells: 42
Some preliminary analyses and step-by-step instructions for downloading, mapping, and visualizing these raw data are described on the Bergman lab blog. This analysis shows that the depth of coverage for this dataset is >90x for reads mapping to autosomes in the D. melanogaster Release 5 reference genome sequence. Dr. Bergman also shows that individual PacBio long reads can uniquely localize repetitive transposable elements up to ~10 kb in size, and can be used to fill at least one of the rare but persistent gaps remaining in the euchromatic portion of the reference genome.
Most assembly algorithms collapse contigs into a single copy of the genome (haploid); however this does not reflect the true underlying state in diploid genomes such as flies or human, which have two copies of each chromosome – inheriting one copy from the mother and another copy from the father. In the case of this inbred subline, with limited allelic variation, both assembly strategies are feasible. We attempted both a traditional haploid assembly as well as a first-ever diploid assembly of the D. melanogaster genome, with preliminary results summarized below. The maximum contig length in both haploid (25 Mb) and diploid (21 Mb) assemblies produced contigs that span almost the entire length of chromosome arm 3L:
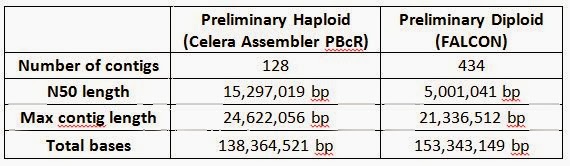
The preliminary haploid assembly using the PacBio Corrected Reads (PBcR) pipeline in Celera® Assembler 8.1 was carried out in collaboration with Drs. Sergey Koren and Adam Phillippy at the University of Maryland. They were able to assemble entire chromosome arms de novo into fewer pieces than the current version of the reference genome (Release 5) – an effort that has spanned two decades, cost millions of dollars, and involved laborious BAC design and sequencing as well as manual finishing and optical mapping. This level of completeness in a de novo assembly is unprecedented in a metazoan genome, and required a total of 6 weeks from initial fly collection and sorting to final analysis and assembly. The actual sequencing time was 6 days. The contig count for each chromosome is tabulated below.
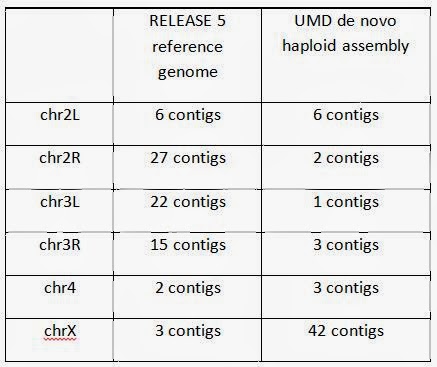
The larger number of contigs for chromosome X can be explained because only adult males were sequenced here (with a 50:50 ratio of chrX to chrY), whereas the Release 5 genome is from mixed-sex embryos. Chromosome X and Y assemblies can further be improved using a higher coverage of reads, and this analysis is underway. Additional analysis and data associated with the haploid assembly are provided here by Dr. Sergey Koren and Dr. Adam Phillippy, including 70x of high-quality, pre-assembled reads using the Celera Assembler PBcR pipeline. Dr. Koren will be presenting the full results at the International Plant and Animal Genome XXII meeting on Tuesday, January 14th.
We also attempted a diploid assembly using an early version of FALCON (Fast Alignment and CONsensus), a new assembly algorithm that is currently being developed at PacBio to explore de novo assembly of diploid genomes. The FALCON assembler extends the Hierarchical Genome Assembly Process (HGAP), with faster implementations of the aligner and consensus algorithms for the pre-assembly and overlap steps of the process. A string-graph is used for the layout stage, which preserves structural and phasing information in polymorphic and heterozygous diploid genomes. The algorithm outputs “primary contigs” and their “associated contigs”, capturing alternative local variants associated with a primary contig. Details were presented at the Genome Informatics Conference in Cold Spring Harbor in November 2013 with slides available here. We assessed the results of our preliminary FALCON assembly by aligning the assembled contigs (light green) to the euchromatic arms (dark green) of the D. melanogaster reference genome using nucmer from the Mummer3 package. Alignments to 2L, 2R, 3L, 3R, 4, X and Y are shown below with sequence identity of each alignment block color-coded in blue indicating >99.96% identity and yellow indicating >99.9% identity. The blue arches represent “unused edges” in the FALCON string graph, and can be interpreted as potential connection points if a more aggressive contig joining threshold were desired.

By sequencing sex-selected male flies, we were able to increase the coverage of reads from the Y-chromosome, which remains one of the most challenging regions left to assemble in the reference genome. Only ~1% of chromosome Y is represented in the reference (Release 5). More recently the BDGP has assembled 7.5% of the Y and we anticipate more than 50% of the Y-chromosome can be assembled with the new data. Ken Wan and Sue Celniker identified contigs containing Y linked genes. The self-self dot-plot below shows that FALCON was able to de novo assemble a 650 kb region of the heterochromatic region of chromosome Y containing a very complex nested repeat structure. In addition, the gene Pp1-Y2 (a testis-specific phosphatase) which spans several gaps of unknown size in the Release 5 sequence is now entirely contained in this single contig:

A development version of FALCON is available on github to developers and bioinformaticians interested in this new assembly process.
We are also releasing an updated dataset of the Arabidopsis thaliana genome using the latest P5-C3 chemistry for comparison to our initial data release using P4-C2 chemistry. In total, we have now released four datasets from three important model organisms: S. cerevisiae (yeast), A. thaliana (flowering plant), and D. melanogaster (fruit fly). This data is freely available and we invite the research community to explore the collection:
S. cerevisiae: P4-C2 Chemistry
A. thaliana: P4-C2 Chemistry, P5-C3 Chemistry
D. melanogaster: P5-C3 Chemistry