We are pleased to make publicly available a new shotgun sequence dataset of long PacBio® reads from a human DNA sample. We previously released sequence data using Single Molecule, Real-Time (SMRT®) Sequencing of ~10x coverage of this sample, sufficient for reference-based detection of structural variation. Today we expand on that release with additional data that increases the total sequencing coverage to ~54x. This long-read data has enabled the generation of the first de novo human genome assembly from PacBio-only sequence reads. Download the 54x long-read coverage dataset.
The dataset was generated from sequencing a well-studied human cell line (CHM1htert), which is being utilized as part of a National Institutes of Health project to sequence and assemble an alternate reference genome (the “platinum genome”). This NIH project is being led by Rick Wilson from Washington University at St. Louis and Evan Eichler from the University of Washington in collaboration with investigators from the National Center for Biotechnology Information.
This new PacBio-only genome assembly marks a continuation of recent data releases highlighting the power of long reads for generating high-quality de novo genome assemblies of increasing size and complexity (Figure 1). For the human genome, it is a follow-on from the October 2013 release of a ~10x coverage dataset for detecting structural genomic variation. Our aim is to help scientists resolve the many structural variants that have been difficult or impossible to characterize using short-read technologies.Identifying these variants, such as large deletions, inversions, and repeat elements, is a prerequisite to understanding many diseases and thereby offers great potential in biomedical research and clinical treatment. Thus, it is essential to have full and accurate representations of these in human genome data. In addition, we believe that higher-quality de novo assemblies of human genomes will enable a greater understanding of genetic variation in genomes at all size scales in a hypothesis-free manner, without bias from conventional reference-guided approaches.
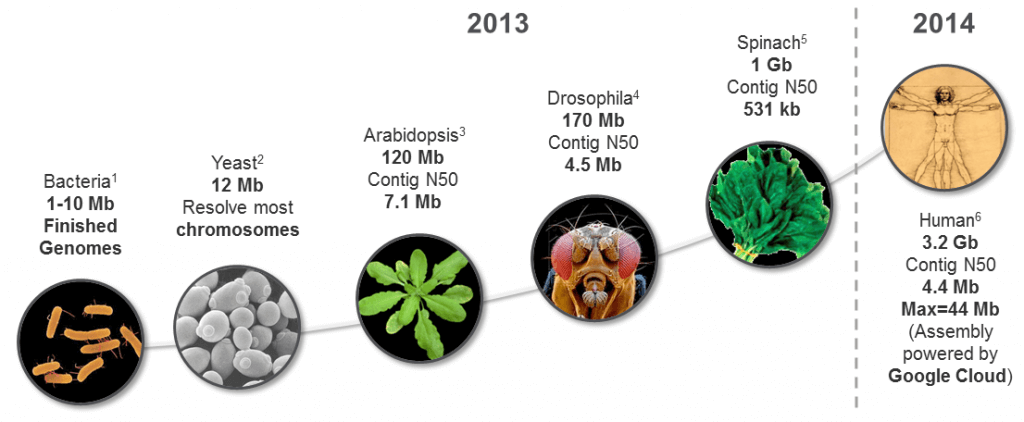
Figure 1. Progress of PacBio-only de novo assembly. (For sources, see References 1-6 below.)
Just one sequencing library type was required for this effort, in the form of ~20 kb long-insert shotgun libraries, which were size-selected using the BluePippin™ platform from Sage Science and sequenced with our P5-C3 chemistry on the PacBio RS II using 180-minute movies.
Below are some sequencing statistics of the dataset:
• Total number of reads: 21,856,161
• Total number of post-filtered bases: 167,851,128,644 bp
• Average throughput/SMRT Cell: 608 Mb
• Average read length: 7,680 bp
• Half of sequenced bases in reads greater than: 10,739 bp
• Longest DNA insert sequenced: 42,774 bp
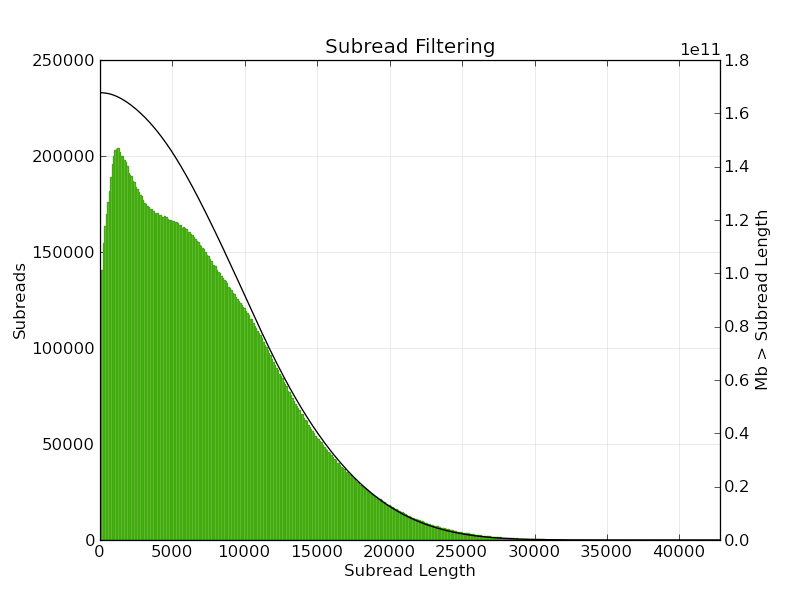
Figure 2. Subread length distribution. A subread is a DNA insert sequenced between two SMRTbell™ hairpin adapters. The solid black line (right y axis) denotes the amount of sequenced bases greater than a given subread length (x axis).
This project also offered opportunities to apply the current Hierarchical Genome Assembly Process (HGAP) tool chain for generating a first PacBio-only de novo assembly of a human genome. This assembly represents the initial result straight out of the assembly pipeline, and we and our collaborators are now working on curating and polishing the assembly. We teamed up with Google to use the Google® Cloud Platform for the most computationally intensive part of the HGAP pipeline. In a single day, the platform executed 405,000 CPU hours to align the long reads to each other. The output alignment data was transferred back to PacBio for generating pre-assembled reads using a modified version of FALCON. We then used Celera® Assembler 8.1 to generate the assembly, and our consensus caller Quiver was applied for the final sequence. The pipeline produced a 3.25 Gb assembly with a contig N50 of 4.38 Mb, and the longest contig of 44 Mb. In comparison, the most recent reference-guided assembly using Illumina® sequencing and BAC-clone finishing on the same sample had a total assembly size of 2.83 Gb and a contig N50 of 144 kb (Figure 3).
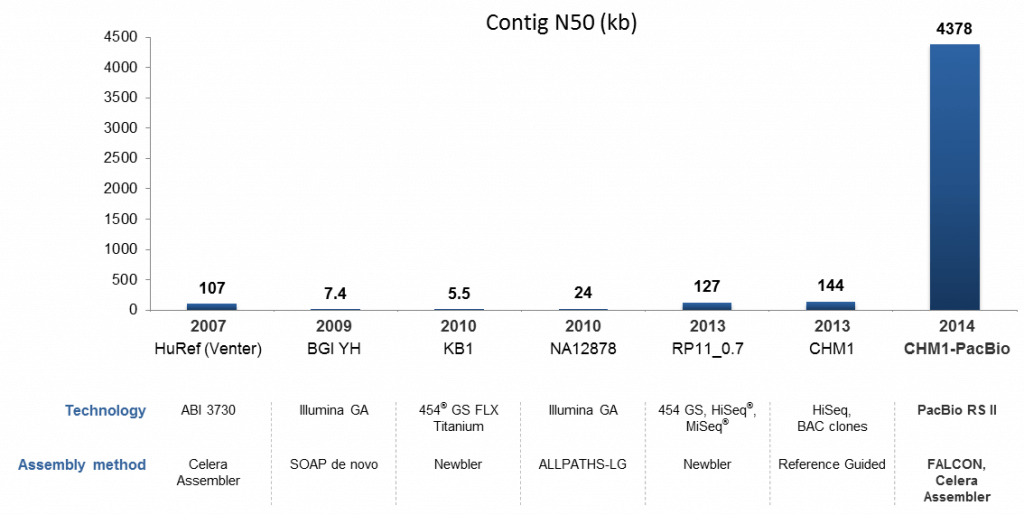
Figure 3. Historical comparison of human genome de novo assemblies including the 2007 HuRef assembly to the 2014 PacBio-only 2014 assembly. Data sources: HuRef (Venter); BGI YH; KB1; NA12878; RP11_0.7; 2013 CHM1.
This project will be highlighted in several presentations at this week’s Advances in Genome Biology and Technology (AGBT) conference, including more details on the assembly process during Jason Chin’s presentation, entitled “String Graph Assembly For Diploid Genomes With Long Reads,” on Friday at 8:30 pm. The data will also be highlighted in the PacBio workshop from our CSO Jonas Korlach on Friday at 2:40 pm. By releasing this dataset, we hope to support the bioinformatics community, along with our own efforts, to further develop and optimize computational algorithms and genome assembly pipelines for large-scale genome assemblies and structural variant detection using SMRT Sequencing. We look forward to the generation of many additional high-quality human genome de novo assemblies to reveal new insights into human genetics.
References
- http://genomebiology.com/2013/14/9/R101
- https://github.com/PacificBiosciences/DevNet/wiki/Saccharomyces-cerevisiae-W303-Assembly-Contigs
- https://github.com/PacificBiosciences/DevNet/wiki/Arabidopsis-P5C3
- https://github.com/PacificBiosciences/DevNet/wiki/Drosophila-sequence-and-assembly
- https://stream.dcasf.com/webinar/a-de-novo-draft-assembly-of-spinach-using-pacific-biosciences-technology/
- http://datasets.pacb.com/2014/Human54x/fast.html