By Jonas Korlach, Chief
Scientific Officer
Single Molecule, Real-Time (SMRT®) DNA sequencing achieves
highly accurate sequencing results, exceeding 99.999% (Q50) accuracy,
regardless of the DNA’s sequence context or GC content. This is possible
because SMRT sequencing excels in all three categories that are relevant when
considering accuracy in DNA sequencing:
highly accurate sequencing results, exceeding 99.999% (Q50) accuracy,
regardless of the DNA’s sequence context or GC content. This is possible
because SMRT sequencing excels in all three categories that are relevant when
considering accuracy in DNA sequencing:
1.
Consensus accuracy
Consensus accuracy
2.
Sequence context bias
Sequence context bias
3.
Mappability of sequence reads
Mappability of sequence reads
Since there has been some confusion in the community
about accuracy in SMRT sequencing, I would like to describe how our system
performs and how such high accuracy is achieved. You can download the full perspective, complete with graphs and figures. What follows below is a summary of that
document.
about accuracy in SMRT sequencing, I would like to describe how our system
performs and how such high accuracy is achieved. You can download the full perspective, complete with graphs and figures. What follows below is a summary of that
document.
Consensus Accuracy
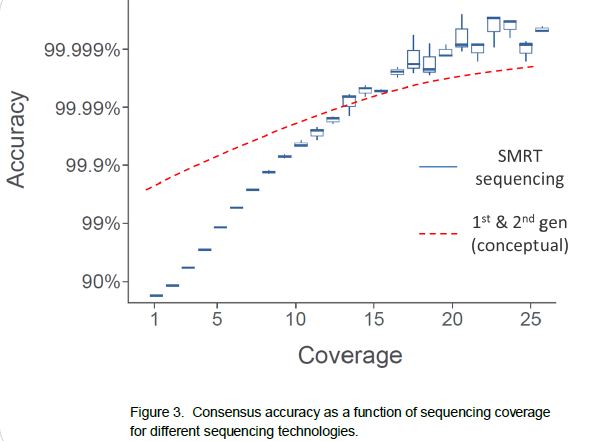
Sequencing results are generated through consensus
analysis, averaging the sequence information from multiple reads for each
reference position. While it is true that single-pass sequence reads in SMRT
sequencing are more error-prone (median error of currently ~11%), it does not
prohibit the determination of a high-quality consensus. We have developed a
mapping tool called BLASR which allows confident mapping of SMRT sequencing reads to their respective
locations in the reference, despite the higher single-pass error rate. Then, as
is the case for second-generation sequencing, results are generated through
consensus, taking reads that map to the same region in the reference and
averaging the base calls for each reference position. A key feature in SMRT
sequencing that allows the determination of a >99.999% accurate consensus
lies in the fact that the single-pass errors are distributed randomly, which
means that they wash out very rapidly upon building consensus. Using the
characteristic features of SMRT sequencing reads, we have developed a consensus tool called Quiver
that delivers the high-quality consensus sequence. Because of the random nature
of the errors, SMRT sequencing in many cases exceeds the accuracy achieved by
other sequencing methods.
analysis, averaging the sequence information from multiple reads for each
reference position. While it is true that single-pass sequence reads in SMRT
sequencing are more error-prone (median error of currently ~11%), it does not
prohibit the determination of a high-quality consensus. We have developed a
mapping tool called BLASR which allows confident mapping of SMRT sequencing reads to their respective
locations in the reference, despite the higher single-pass error rate. Then, as
is the case for second-generation sequencing, results are generated through
consensus, taking reads that map to the same region in the reference and
averaging the base calls for each reference position. A key feature in SMRT
sequencing that allows the determination of a >99.999% accurate consensus
lies in the fact that the single-pass errors are distributed randomly, which
means that they wash out very rapidly upon building consensus. Using the
characteristic features of SMRT sequencing reads, we have developed a consensus tool called Quiver
that delivers the high-quality consensus sequence. Because of the random nature
of the errors, SMRT sequencing in many cases exceeds the accuracy achieved by
other sequencing methods.
Sequence Context
Bias
Bias
Many sequencing systems have difficulties sequencing
through extremely AT-rich or GC-rich DNA regions, highly repetitive sequences, palindromic
sequences, or long homonucleotide stretches, either yielding no sequence or
sequence of poor quality. SMRT sequencing does not exhibit such sequence
context bias and performs very uniformly through regions previously considered
difficult to sequence. This advantage has been used to close gaps in genomes
that were called ‘hard stops’ for other sequencing systems. An extreme example
for demonstrating the lack of bias in SMRT sequencing was demonstrated in a
recent paper
by sequencing through thousands of bases of 100% GC content: CGG trinucleotide
repeat expansions which cause fragile X syndrome. Similarly, because there is
no denaturation step in the sample preparation, palindromes can be sequenced
without difficulty.
through extremely AT-rich or GC-rich DNA regions, highly repetitive sequences, palindromic
sequences, or long homonucleotide stretches, either yielding no sequence or
sequence of poor quality. SMRT sequencing does not exhibit such sequence
context bias and performs very uniformly through regions previously considered
difficult to sequence. This advantage has been used to close gaps in genomes
that were called ‘hard stops’ for other sequencing systems. An extreme example
for demonstrating the lack of bias in SMRT sequencing was demonstrated in a
recent paper
by sequencing through thousands of bases of 100% GC content: CGG trinucleotide
repeat expansions which cause fragile X syndrome. Similarly, because there is
no denaturation step in the sample preparation, palindromes can be sequenced
without difficulty.
Mappability of
Sequence Reads
Sequence Reads
Even if a sequence read is
100% accurate, it can still be uninformative or even misleading if it cannot be
mapped correctly onto the reference genome. If a read is not long enough to
span a repetitive region in the genome with at least one unique flanking
sequence, the origin of the read cannot be determined unequivocally, and thus
any variation observed in this read is ambiguous with respect to where this
variation occurred in the genome. The long SMRT sequencing reads avoid
mismapping by providing long, multi-kilobase reads that can stretch through
repetitive genomic regions and anchor those reads to their correct location.
Summary
Through the combination of high consensus accuracy due to
a random error profile, lack of sequence context bias, and very long reads that
avoid mismapping artifacts, SMRT sequencing produces comprehensive and highly accurate
sequencing results. The higher rate of single-pass errors is not a problem
because they wash out rapidly when building consensus. It is important to use
appropriate bioinformatic tools that take into account the specific
characteristics of SMRT sequencing to obtain the highest quality results. With
these characteristics, we strive to generate the highest possible scientific
value for each base sequenced – in its native, multi-kilobase context – to the
scientific community.
a random error profile, lack of sequence context bias, and very long reads that
avoid mismapping artifacts, SMRT sequencing produces comprehensive and highly accurate
sequencing results. The higher rate of single-pass errors is not a problem
because they wash out rapidly when building consensus. It is important to use
appropriate bioinformatic tools that take into account the specific
characteristics of SMRT sequencing to obtain the highest quality results. With
these characteristics, we strive to generate the highest possible scientific
value for each base sequenced – in its native, multi-kilobase context – to the
scientific community.