It seems like there is a new story every week in the mainstream press about the unexpected ways the bacteria living on and within us impact health, disease, and even our behavior. The torrent of new discoveries unleashed by high-throughput sequencing has captured the imagination of scientists and laypeople alike. Scientists at Second Genome are hoping to apply these insights to improve human health, leveraging their bioinformatics expertise to mine bacterial communities for potential therapeutics. Second Genome is a clinical stage pharmaceutical company with a mission to redefine disease in the context of microbiome medicine and create therapeutics that can address unmet medical needs. Recently they teamed up with scientists here at PacBio to explore how long-read sequencing might supplement their short-read-based pipeline for gene discovery, using an environmental sample as a test case. They were especially interested in identifying unique, complete, and error-free gene clusters in metagenomic assemblies.
The team began by spiking the sample with internal controls and generating a 10 kb insert library. The library was sequenced on two SMRT Cells with 20-hour movies on the Sequel System. The resulting data was analyzed in two ways. First, the ccs algorithm was applied to generate ~270,000 HiFi reads per 1M SMRT Cell @ 99% minimum accuracy. Since these reads are on average 10 times longer than the average bacterial gene, full-length genes can be discovered even without assembly. Bioinformaticians at Second Genome then used two different gene prediction programs to evaluate the usability of HiFi reads for gene discovery. Next, the raw data was assembled with Canu, and the discovered genes were mapped back onto the contigs.
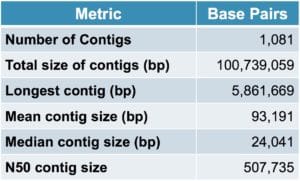
How did the results shape up? As you can see, two SMRT Cells of data revealed an impressive number of genes and a highly contiguous assembly with a mean contig size of 93 kb. In addition, among the contigs were two closed bacterial genomes.
Table 1. Metagenome assembly statistics
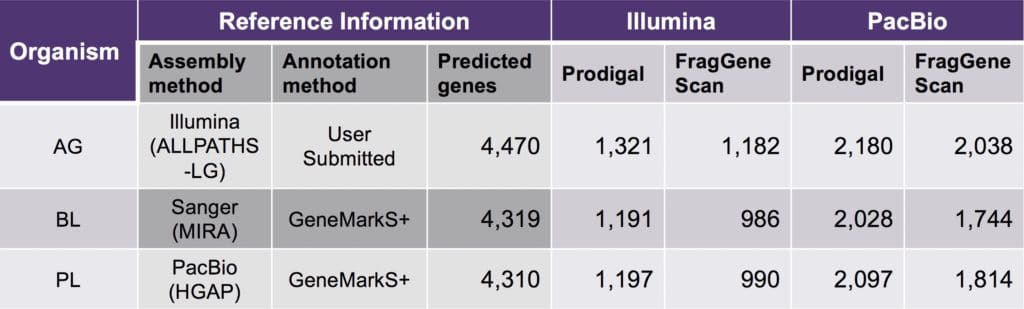
Comparing the performance of the two gene discovery algorithms, scientists at Second Genome found Prodigal predicted a large number of genes that were concordant with expected genes regardless of the sequencing technology, assembler, or annotation tool used in the source genome. However, more analysis is needed to make a conclusion about the program performance.
Table 2. Comparison of gene discovery programs for all identified genes

Second Genome then took the analysis one step further and assessed whether long-read sequencing was a good value for their business. Rather than fall back on the commonly used ‘cost per base’ metric, they developed their own more pertinent way of measuring success: what was the cost per error-free, unique predicted protein? By normalizing their data to calculate unique predicted protein per $1,000, they found that PacBio sequencing was actually twice as cost-effective as short-read technology at discovering complete genes from the same DNA sample. Whereas short-read technology predicted ~17,000 full-length proteins per $1,000 of data, PacBio data yielded ~36,000 predicted proteins. Similarly, PacBio sequencing recovered approximately twice as many spike-in sequences per $1,000 invested.
Table 3. Comparison of yield per $1,000 for different sequencing and gene-calling methods of identifying full-length proteins from spike-in genomes

The results demonstrate that long-read sequencing technology can be successfully applied to metagenomes from complex communities and is complementary to short-read technology. PacBio sequencing was more effective at discovering complete genes than short-read sequencing in this study. Even more exciting is the prospect of replicating this study on the forthcoming Sequel II System which, with eight times as many ZMWs, will make the cost comparison even more favorable. Todd DeSantis said, “We anticipate the Sequel II System may enable scientists to more completely characterize microbial communities. This could ultimately yield significant discoveries in the microbiome and metagenomic space at a reduced cost to what we are
seeing today with Sequel and short reads.”