
We are energized from celebrating a Bright New Day at PAG last week! We had a fantastic time connecting with the plant and animal genomics community through collaborative discussions at our booth and sharing our passion for accelerating advancements in agriculture and biodiversity at our industry workshop. Now reflecting on the important themes of the conference, we have been thinking about the ever-pressing need to improve crop production in order to meet the demands of a rapidly growing global population.
HiFi sequencing technology is already proving to be a game-changing tool to address this challenge for breeding programs. Highly accurate HiFi long reads enable precise discovery of not only single nucleotide polymorphisms (SNPs) but also larger structural variants (SVs), which can be more closely tied to traits of interest within a population. To catalog all variants within a species, researchers have increasingly turned to HiFi sequencing to generate more complete and accurate reference genomes and pangenomes. Agrigenomics researchers are leveraging these capabilities to improve crop breeding by identifying and selecting high-value traits faster and with greater accuracy, ultimately addressing challenges like disease resistance, drought tolerance, and yield improvement.
Now, the emergence of low-pass HiFi sequencing offers more than 8x the cost efficiency of short reads while retaining the key advantages of long reads, making accessible high-resolution trait mapping a reality.
A new approach to trait mapping
A preprint out just last week entitled “Long-read low-pass sequencing for high-resolution trait mapping,” highlights the potential for this tool in plant breeding. In this study, researchers from HudsonAlpha and the University of Georgia compare the performance of HiFi sequencing against Illumina short reads for low-pass trait mapping of the peanut (Arachis hypogaea) genome. Using both a single reference genome and a pangraph, the authors use the Khufu analysis pipeline, now made commercially available by authors Josh Clevenger, Alex Harkess, and Kendall Lee through Veil Genomics, to identify and associate genomic variants with traits of interest. With its highly complex tetraploid genome, the peanut serves as an ideal test case for low-pass HiFi sequencing in trait mapping.
Greater clarity for traits of interest
The researchers employed a high-throughput, low-input protocol for sequencing on the Revio system. At an average sequencing depth of 1.63x, HiFi sequencing covered 55% of the genome and 58% of gene space, significantly outperforming short-read low-pass sequencing at 1.68x depth, which achieved only 17% and 11%, respectively. HiFi reads demonstrated 92.9% confidence in alignment, far surpassing the 54.9% retention rate observed for short reads. The authors stress that this increased alignment confidence enables a more comprehensive view of repetitive and complex genomic regions, which are often difficult to analyze with short reads.
Using their newly developed Khufu analysis pipeline and a 16-genome pangraph, this study revealed that HiFi reads identified approximately 18.7 times more SNPs, 45.9 times more indels, and 51.6 times more structural variations than short reads. The average lengths of insertions and deletions, which made up the majority of the “high impact” variants disruptive to protein coding, were 174 bp and 1246 bp, respectively. Both of these variant lengths are longer than the average short read length, meaning they would not have been captured with short-read sequencing.
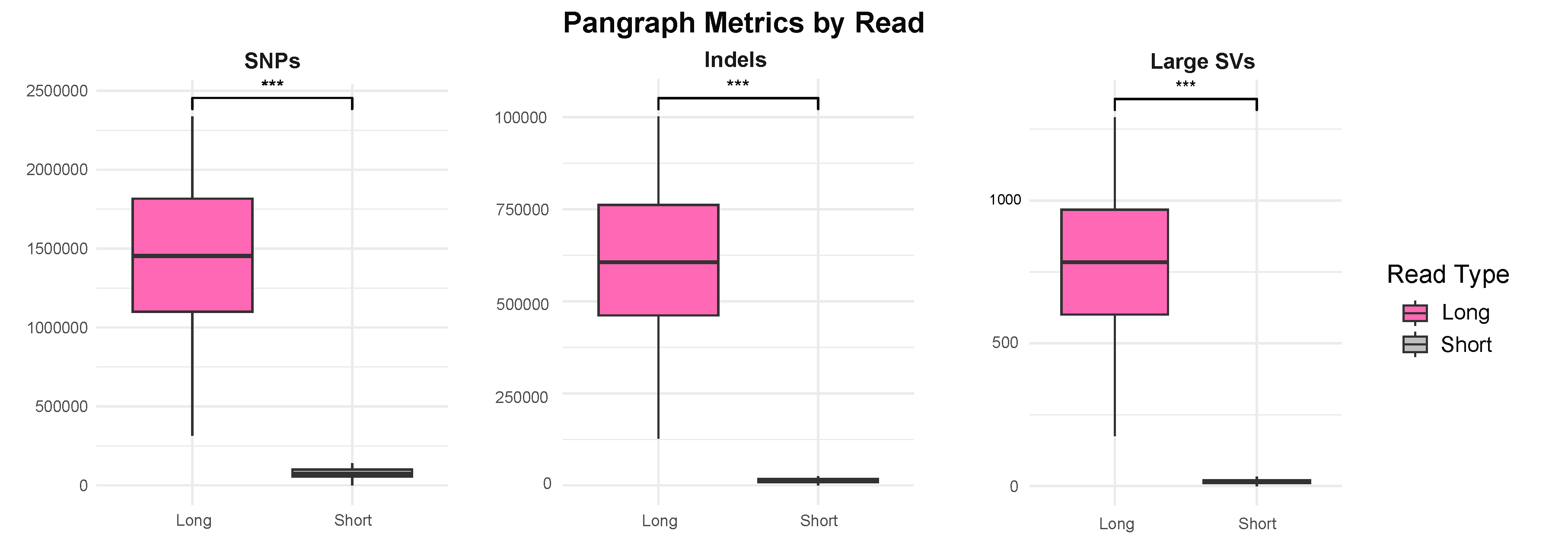
This comprehensive variant detection is essential for understanding the genetic basis of key traits. Furthermore, regardless of sequencing depth, HiFi sequencing consistently produced higher locus similarity scores for disease resistance loci linked to late leaf spot and tomato spotted wilt virus, emphasizing the enhanced accuracy of genomic selection when using long reads versus short reads.
Lower “cost per insight”
The authors also conducted a cost-effectiveness analysis and conclude that “long reads provide significantly greater genomic insight per unit cost” at an average of over 8 times the cost efficiency of short reads.

“These findings position long-read sequencing as a more economical choice for high-resolution genomic analyses, particularly in projects requiring extensive coverage of complex genomes.” -Lee et al. 2025
Enabling breakthroughs in crop breeding
High-resolution genomic data is key to accurately selecting traits like drought tolerance, disease resistance, and yield improvement that are critical for sustainable crops. Traditional short-read sequencing can often fall short in capturing these contributing variants, particularly at low coverage. By contrast, the ability of HiFi sequencing to span large and complex regions of the genome ensures more accurate and comprehensive variant detection at any depth.
By addressing one of the major bottlenecks in plant breeding—the ability to efficiently and accurately map traits to markers—low-pass HiFi sequencing represents a paradigm shift in how breeders approach crop improvement. This method makes it easier to identify the genetic foundations of key traits and incorporate them into commercial cultivars. And with increased cost efficiency, low-pass HiFi sequencing enables researchers and breeders to scale up their efforts, accelerating the development of crops with improved resilience, higher yields, and better adaptability to environmental challenges.
A sustainable future for food
This study reinforces the power of low-pass HiFi sequencing as a cost-effective alternative to short-read sequencing for high-resolution trait mapping. At equivalent coverage, long reads far exceed short reads in confidently calling SNPs, identifying high-value variants, and capturing the broader variant landscape. These capabilities make low-pass HiFi sequencing an indispensable tool for modern plant breeding, helping researchers and breeders overcome challenges in trait selection and accelerate the development of resilient, high-yield crops.
As global agriculture faces mounting pressures, innovative approaches like low-pass HiFi sequencing provide hope for a more secure and sustainable food future. PacBio is proud to support this groundbreaking research and remains committed to advancing genomic technologies that drive meaningful change in agriculture.
To learn more about using low-pass HiFi sequencing with your workflows, contact Veil Genomics or PacBio.
Explore PacBio sequencing solutions for agrigenomics with this brochure and at our website.
References
Lee, K. C., et al. (2025). Long-Read Low-Pass Sequencing for High-Resolution Trait Mapping. bioRxiv. https://doi.org/10.1101/2025.01.09.632261