UPDATE:
Our R&D team has added a new dataset for the MCF-7 human breast cancer transcriptome, originally released in 2013. The new results were produced using 28 SMRT® Cells with 4-hour movies and P5-C3 chemistry. Sizing was performed with the SageELF™ platform (fractions collected: 1-2 kb, 2-3 kb, 3-5 kb, and 5-10 kb). Sequencing of the larger fractions with our newer sequencing chemistry that generates longer reads added longer transcripts (up to 10 kb) to the MCF-7 dataset, which previously had only transcripts up to 4 kb.
New FASTA and GFF files are available, representing the new combined dataset. Raw data for both the 2013 and 2015 sequencing is also available.
ORIGINAL POST (December 11, 2013):
Understanding the biology of a genome requires knowing the full complement of mRNA isoforms. In recent years, microarrays, high-throughput cDNA sequencing, and RNA-seq have become very useful tools for studying transcriptomes. High-throughput cDNA sequencing is accurate but laborious, while the inherently complex nature of the transcriptome makes transcript assembly computationally intractable. Recently, Steijger et al. (1) showed that complete isoform reconstruction from RNA-seq short-read data remains challenging even when all constituent exons are identified.
A number of recent publications have demonstrated the utility of full-length transcript sequencing by taking advantage of the long read lengths of SMRT® Sequencing technology (2)–(4). SMRT Sequencing produces reads that originate from independent observations of single molecules; no assembly is needed if a read spans the entire length of the transcript. To demonstrate the capabilities of PacBio® Isoform Sequencing (Iso-Seq) technology and show a glimpse of the complexity of eukaryotic transcriptomes, we generated a deep dataset of full-length cDNA sequencing of RNA from MCF-7, a human breast cancer cell line. The sequencing data was collected from several internal training sessions where different library preparation techniques were tested. We are releasing the underlying data in an effort to aid the design of future PacBio Iso-Seq experiments and to spur advances in the development of bioinformatics tools for analyzing full-length transcripts.
In our final dataset, we obtained 44,531 non-redundant transcript-length consensus sequences ranging from 400 bp – 4,900 bp, with an average length of 1,929 bp (Fig. 1a). The total percentage of consensus bases that disagreed with the hg19 genome is 0.27%, out of which 0.16% are due to substitutions and thus could likely be true SNPs (Fig. 1b). About half of the transcribed loci have one observed isoform, while the rest have mostly 2-5 isoforms (Fig. 2). We compared our predicted full-length transcripts against the known annotations and found that we were able to recover full-length alternative splice forms (Fig. 3), alternative polyadenylation, novel transcripts, and known fusion genes (Fig. 4). We encourage interested researchers to explore the dataset.
Materials & Methods
Full-length cDNA was generated from polyA RNA using standard cDNA synthesis kits (Clontech® SMARTer™ and Invitrogen® Superscript® kits). To capture longer, rarer transcripts in sufficient abundance, parts of the double-stranded cDNA were size selected into three fractions, which were subsequently amplified and converted into SMRTbell™ templates. Details on the sample preparation can be found on Sample Net. SMRTbell libraries were sequenced using the P4-C2 sequencing chemistry with 2-hour movies.
After sequencing, we computationally determined the completeness of the sequences using polyA-tail signals and library adapters. To obtain a non-redundant set of full-length, high-quality transcript sequences without bias from other sequencing platforms, we developed a de novo, isoform-level clustering algorithm that uses only PacBio data. Briefly, the algorithm iteratively clusters reads to generate consensus sequences that represent the original transcripts. The algorithm takes into account the existence of the polyA-tail signal to differentiate isoforms with alternative stop sites. The final consensus sequences were called using Quiver and filtered to create the final polished, full-length, non-redundant dataset. Details of the clustering algorithm will be described in two upcoming webinars on Wednesday, January 22 at 8 AM PST and 5 PM PST.
Some statistics from the sequencing and results are listed below:
- Number of SMRT Cells: 119
- no-size selection: 12
- 1-2 kb: 37
- 2-3 kb: 37
- > 3 kb: 33
- Total number of post-filtered bases: 14,062,161,755

Figure 1. (a) Length distribution of polished, non-redundant transcript sequences. Each transcript sequence represents a unique isoform. (b) Breakdown of differences to hg19. Consensus sequences were mapped to hg19 using GMAP (version 2013-07-20) with default parameters. Different error categories were aggregated over all 44,531 transcript sequences. Some errors are likely to be due to real biological differences from the reference sequence.

Figure 2. Number of isoforms per loci. Transcripts that overlap on the genomic coordinate by 1 bp are grouped together to form non-overlapping transcribed loci. Total number of loci: 14,385. The majority (61%) of transcribed loci have only 1 or 2 transcripts while 0.6% of them have 20 or more isoforms. This is consistent with other studies of full-length cDNA sequencing of a single sample type [5].
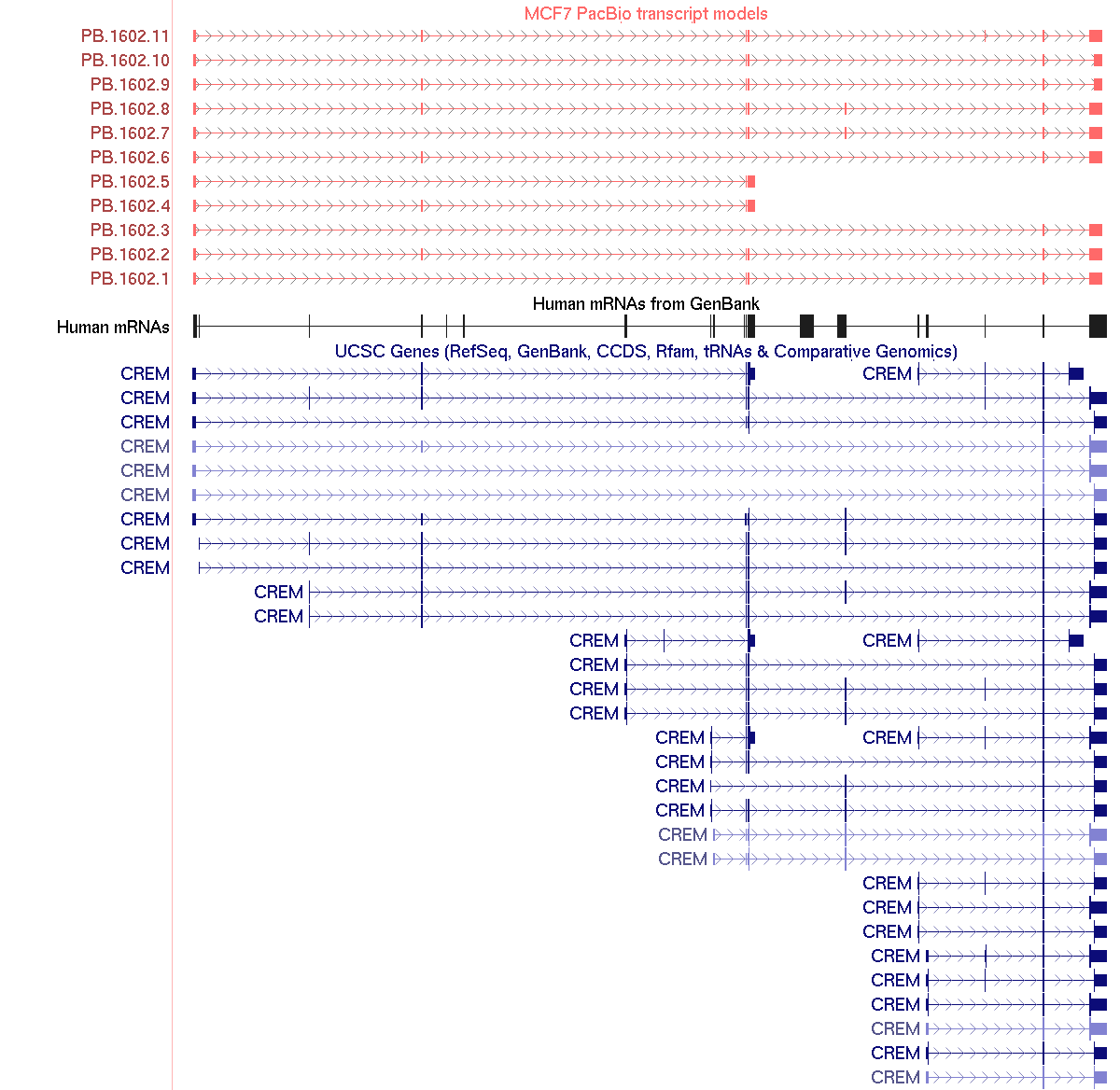
Figure 3. UCSC browser screenshot of the CREM gene region. PacBio transcripts (top, red) capture multiple isoforms of the CREM gene, including alternatively spliced exons and alternative poly adenylation sites.
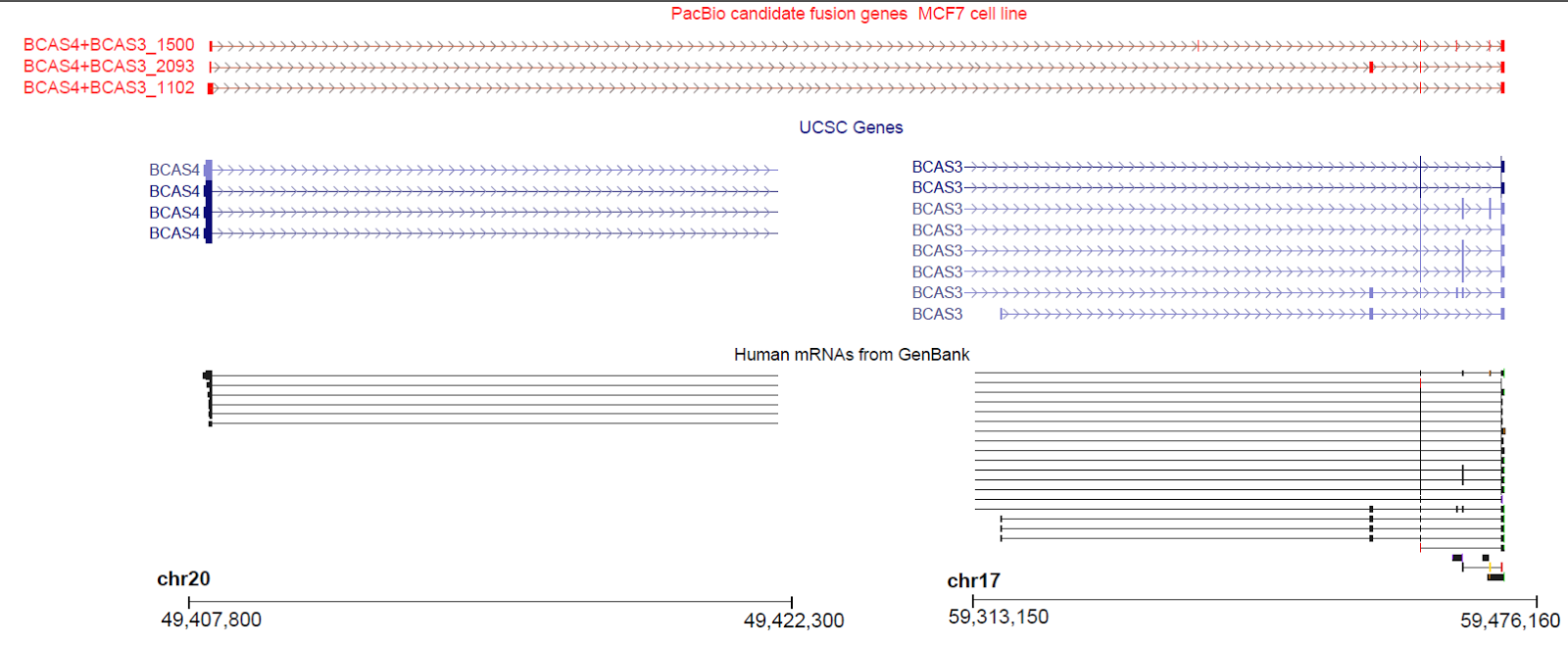
Figure 4. Known cancer fusion gene BCAS4/BCAS3 identified. PacBio transcripts (top, red) show three different fusion variants of the BCAS4/BCAS3 genes. All three variants contain a portion of the 5’ region of the BCAS4 gene (chr20q13) and a portion of the 3’ region of the BCAS3 gene (chr17q23).
We welcome researchers to download and use the dataset for their research. For citation of the dataset, please use:
References
- T. Steijger, J. F. Abril, P. G. Engström, et. al., “Assessment of transcript reconstruction methods for RNA-seq,” Nat. Methods, vol. 10, no. 12, pp. 1177–1184, Nov. 2013.
- D. Sharon, H. Tilgner, F. Grubert, and M. Snyder, “A single-molecule long-read survey of the human transcriptome,” Nat. Biotechnol., vol. 31, no. 11, pp. 1009–1014, Nov. 2013.
- W. Zhang, P. Ciclitira, and J. Messing, “PacBio sequencing of gene families-a case study with wheat gluten genes,” Gene, 2013.
- K. F. Au, V. Sebastiano, P. T. Afshar, J. D. Durruthy, L. Lee, B. A. Williams, H. van Bakel, E. E. Schadt, R. A. Reijo-Pera, J. G. Underwood, and W. H. Wong, “Characterization of the human ESC transcriptome by hybrid sequencing,” Proc. Natl. Acad. Sci. U. S. A., Nov. 2013.