HiFiViral COVID-19 surveillance
Introducing HiFiViral for SARS-CoV-2 kit: a simple-to-use, scalable, cost-effective solution for sequencing the entire SARS-CoV-2 genome. This kitted solution provides robust performance on novel variants and comprehensive detection of all types of mutations.

Benefits for pathogen surveillance

Simple to use
The PacBio HiFiViral kit has less pipetting and fewer plate transfers than PCR-based enrichment assays. The add-only enrichment workflow further reduces handling errors with color-change indicators at every step.

Robust performance
Our fully kitted solution uses a MIPs-based method that combines the simplicity and cost effectiveness of PCR with the variant-robustness of deeply tiled target capture methods. Each base is covered by 22 probes.

Capture all variants
The combination of 675 bp inserts and the accuracy of HiFi sequencing enables comprehensive detection of all types of mutations and the ability to detect major and minor strains.

Cost-effective
PacBio HiFi sequencing on the Sequel II systems is a cost-effective solution that provides built-in surge capacity to scale throughput on a single platform.
Spotlight
Illustrating the new workflow
This short video describes the simplicity of the HiFiViral for SARS-CoV-2 kit solution, which will streamline your workflow and enable you to capture all variants, leading to the best quality results in your SARS-CoV-2 surveillance.
BLOG
Learn how other scientists have used HiFiViral SARS-CoV for COVID-19 research and surveillance:
- Introducing first fully-kitted solution for SARS-CoV-2 surveillance and microbial whole genome assembly with HiFi sequencing
- Omicron and beyond: HiFiViral Kit provides labs with a future-proof solution for emerging COVID-19 variants
- NIH Scientists Chart SARS-CoV-2 Evolution Within an Individual Over Time
- Pandemic Preparedness: PacBio and Labcorp Team Up On a Global Pan-Pathogen Surveillance Network
- For pandemic surveillance, Labcorp scientists sequence thousands of SARS-CoV-2 samples with HiFi reads
HiFiViral for SARS-CoV-2 workflow


Multiplex 24–384 samples per SMRT Cell 8M, or load up to 8 SMRT Cells per Sequel IIe system to process up to 3,072 samples per week.

Sequence and analyze overnight with one touch using SMRT Link v10.2 or later to produce submission-ready files, along with a performance summary and QC tools.
Spotlight
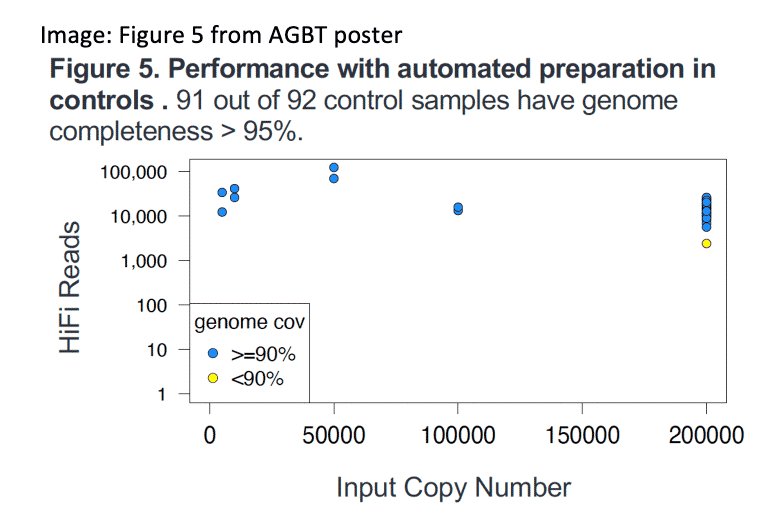
AUTOMATING THE HIFIVIRAL SARS-COV-2 WORKFLOW
Multiple options for automating the HiFiViral SARS-CoV-2 enrichment workflow further improve the scalability of this COVID-19 surveillance assays in a public health environment. In addition, new data shows that HiFiViral continues to return complete viral genome coverage for all circulating strains, including Omicron variants, without probe updates.
Kingan, et. al. (2022) HiFiViral SARS-CoV-2: A mutation tolerant, fully kitted solution for COVID-19 surveillance. Poster presentation, AGBT General Meeting, Orlando, Florida.
{kind=link}
Spotlight
HifiViral SARS CoV2 kit enables genomic surveillance of Covid 19
The HiFiViral SARS-CoV-2 kit is an end-to-end solution for genomic surveillance of COVID-19 variants. The kit utilizes molecular inversion probes (MIPs), a differentiated enrichment technology which combines the simplicity and cost-effectiveness of PCR with the resilience to mutations of target capture. The MIPs approach, coupled with PacBio HiFi reads and SMRT® Link software analysis, delivers several advantages over PCR amplicon sequencing.

Application Brief
HIFIVIRAL SARS-COV-2 KIT FOR COVID-19 WHOLE GENOME SEQUENCING — BEST PRACTICES
HiFiViral for SARS-CoV-2 is a simple-to-use, scalable, cost-effective solution for sequencing the entire SARS-CoV-2 genome. This fully kitted solution uses a novel approach that is robust to new variants and comprehensively detects all types of mutations.