Directly detect epigenetic modifications with HiFi sequencing
HiFi sequencing directly detects DNA modifications in native DNA from variations in the polymerase kinetics of DNA base incorporation, without the need for arduous chemical conversion protocols like bisulfite treatment.
- Detect genome-wide m6A and m4C R-M system motifs at coverage levels recommended for assembly
- Obtain complete genomes with annotations for epigenetic modification
- Reveal phase variation of R-M genes that regulate batteries of genes involved in pathogenesis, host adaption, and antibiotic resistance
Learn how other scientists have used SMRT sequencing to connect prokaryotic methylation to new biology:
- PacBio-powered analysis illustrates how methylation makes the TB pathogen so wily
- SMRT sequencing enables discovery of the epigenetic driver of C. diff persistence
- Creating an epigenetic barcode to accurately characterize microbial communities
Workflow: from DNA to microbial methylome

Sample + library preparation
- Start with unamplified genomic DNA
- Prepare SMRTbell libraries following the protocol for microbial whole genome sequencing.

Sequencing
Data analysis
- Use SMRT Link Microbial Genome Analysis to assemble and annotate active R-M system motifs
- View detailed reports on modifications and associated motifs
Spotlight
BACTERIAL METHYLATION PROFILES HELP ASSIGN ANTIBIOTIC-RESISTANCE GENES TO HOST ORGANISMS IN MIXED ENVIRONMENTAL SAMPLES
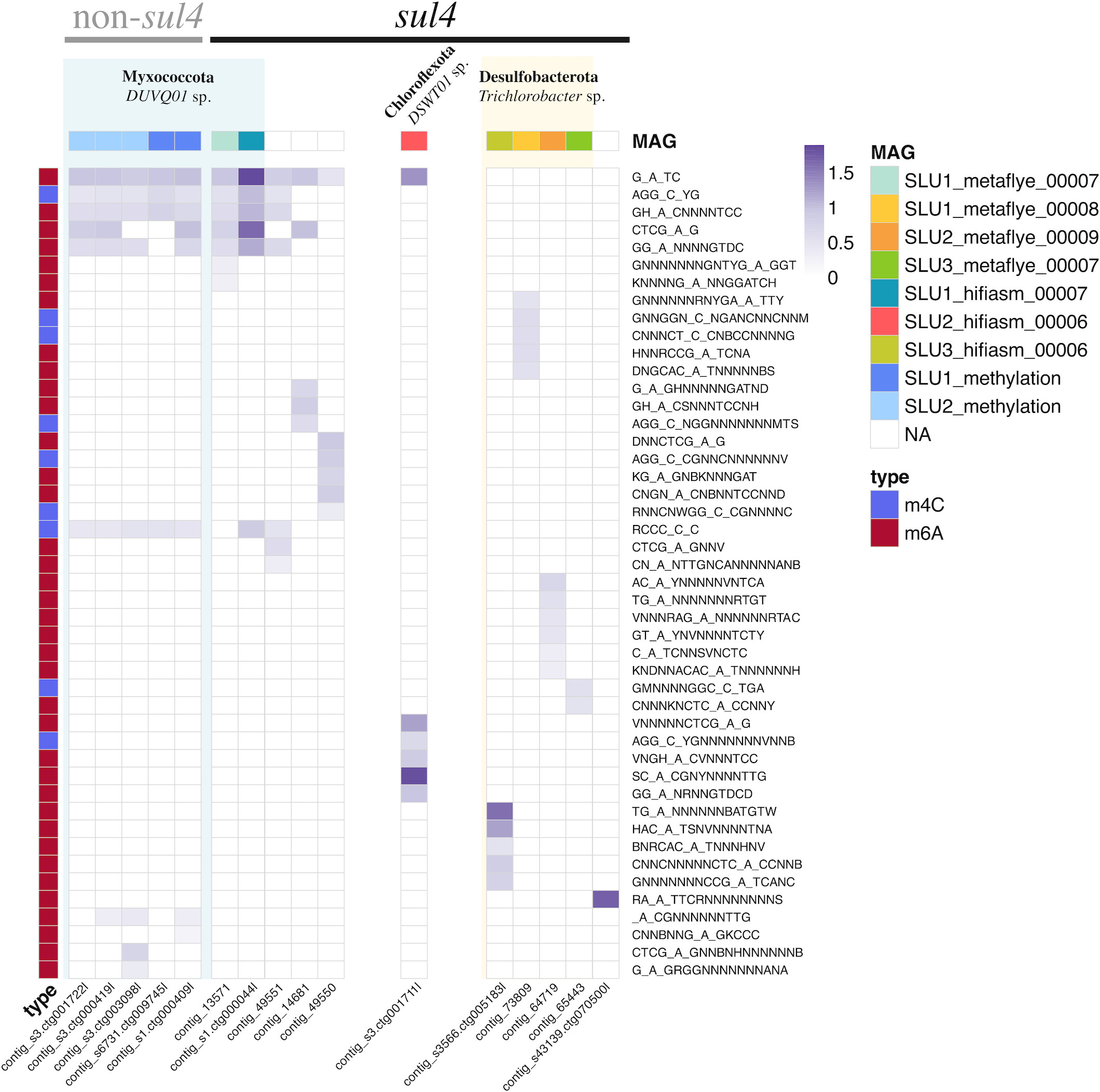
This study used PacBio long-read metagenomics plus bacterial methylation signatures to connect sul4-containing contigs to likely host genomes in complex wastewater communities, grounded in the observation that bacterial taxa carry species-specific methylation patterns because their methyltransferase repertoires differ. For the 12 sul4 contigs, they found an average of 6 methylated motifs of m6A or m4C per contig, with a range of 1 to 13. All but one contig had at least one methylated motif. The methylation patterns grouped the sul4 contigs into three broad categories, which suggested multiple distinct bacterial hosts.
Markkanen, M., et. al. (2026) Sulfonamide resistance gene sul4 is hosted by common wastewater sludge bacteria and found in various newly described contexts and hosts. Microbiology Spectrum. 14:e00857-25, https://doi.org/10.1128/spectrum.00857-25

Spotlight
A role for epigenetics in the regulation of replication and transcription
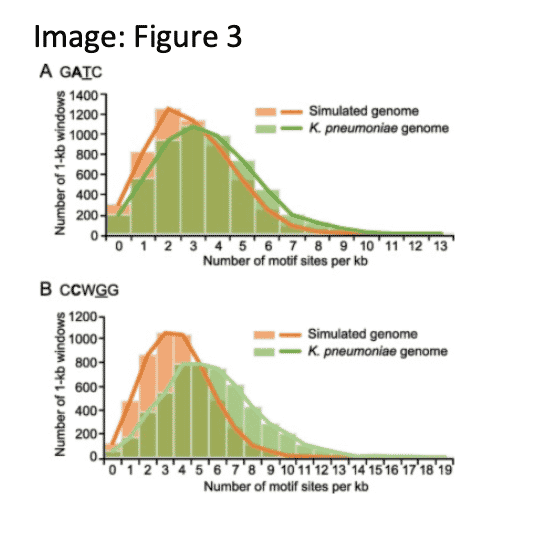
Comprehensive characterization and comparison of the methylomes of 14 K. pneumoniae strains revealed differential concentration of GATC motifs and methylation penetrance in intergenic regions. Modelling of MTase-catalysed methylation suggests slower rates of motifs in replication origins and IGRs, implicating a role in the initiation of replication and transcription.
Fu, J., et. al. (2022). Precision Methylome and In Vivo Methylation Kinetics Characterization of Klebsiella pneumoniae. Genomics, proteomics & bioinformatics, 20(2), 418–434. https://doi.org/10.1016/j.gpb.2021.04.002

{kind=link}
{kind=link}