Whole genome sequencing analysis
Generate exceptional de novo assemblies and uncover the most complete view of genomic variation.

Comprehensive genome assemblies and variant calling
Our whole genome sequencing analysis solutions allow you to choose between easy to use push-button applications or flexible command line tools to generate gold-standard reference genomes, phase haplotypes and call all variant types. Explore the whole genome sequencing application and workflows.
De novo assembly
Generate reference-quality de novo assemblies with megabase-size contig N50s, consensus accuracies >99.99%, and phased haplotypes using highly accurate long reads (HiFi reads).
SMRT Analysis:
Use Genome Assembly to generate high quality (>Q50) phased de novo assemblies, using HiFi reads generated with the circular consensus sequencing (CCS) algorithm:
- An easy-to-use assembler accessible via the SMRT Link graphical user interface that performs fast de novo assembly and phasing of resulting contigs
- Genome Assembly is powered by the improved phased assembly (IPA) algorithm
How it works: Genome Assembly is optimized for HiFi data. Read-to-read overlapping, haplotype phasing, string graph construction, and polishing produce high-quality, haplotype-phased contigs. Haplotype deduplication is included for better haploid representation of the final assembly.

- Convert input to a compressed database for fast and easy retrieval
- Overlap reads
- Phase the overlapped reads
- Remove chimeras and duplicate reads to improve contiguity and assembly quality
- Construct a string graph; extract primary contigs and haplotigs
- Polish the contigs and haplotigs using phased reads
- Identify potential haplotype duplications for better haplotype separation of the final assembly — beneficial for high-heterozygosity samples
Learn more about SMRT Analysis
PacBio DevNet tools
Several options enable assembly of HiFi reads at the command line. PacBio supported tools include pbipa, which allows for automated de novo assembly using IPA. Community-supported tools provide scientists with easy access to open-source analysis tools for de novo assembly and phasing, such as:
HiCanu
Accurate assembly of segmental duplications, satellites, and allelic variants from HiFi reads
Hifiasm
A fast, haplotype-resolved genome assembler
Peregrine
A fast genome assembler for accurate long reads
For assemblies of continuous long reads (CLR) use HGAP4 analysis application
SMRT compatible analysis products
Generating a high-quality genome assembly with PacBio means having access to an end-to-end solution. From cloud-based platforms for running your own analyses to no-hassle full-service genomics, our analysis partners provide many solutions tailored to individual customers’ needs.
DNAnexus
Cloud-based informatics platform providing data analysis tools and assembly services including HGAP, FALCON, and FALCON-Unzip
Computomics
Custom analysis services including de novo assembly of reference-quality genomes
See our complete list of SMRT compatible analysis products
Variant detection
Explore the power of the HiFi reads to discover small variants and phase haplotypes utilizing industry standard analysis tools.
SMRT Analysis
Generate HiFi Reads with the CCS analysis application and align them to a reference sequence with the Mapping application in SMRT Link. Call variants with open source tools such as DeepVariant or GATK, and phase variants with WhatsHap. Visualize results with the integrative genomics viewer (IGV).
How it works: The reads are mapped to the reference sequence then variants are called.


Sequel II system data release: HG002 SV and SNVs (HiFi reads powered by CCS)
Learn more about SMRT Analysis
PacBio DevNet tools
There are several community-supported tools for variant calling and haplotype phasing using SMRT sequencing data. They provide scientists with easy access to additional open-source algorithms, such as:
DeepVariant
DeepVariant is an analysis pipeline developed by Google that uses a deep neural network to call genetic variants from next-generation DNA sequencing data
GATK
Developed in the Data Sciences platform at the Broad Institute, the toolkit offers a wide variety of tools with a primary focus on variant discovery and genotyping
WhatsHap
A software for phasing genomic variants using DNA sequencing reads, also called read-based phasing or haplotype assembly
See our complete list of PacBio DevNet tools for variant detection
SMRT compatible analysis products
Our analysis partners offer several solutions for variant calling. Options include customized analysis based on project needs, full-service genomics analytics, cloud-based platforms with open source tools, or opportunity to run your own algorithms.
DNAnexus
Cloud-based genome informatics platform that provides data analysis tools for variant calling and phasing using GATK, DeepVariant, WhatsHap, and more
Computomics
Custom genome analysis services including variant calling
See our complete list of SMRT compatible analysis products
Structural variant detection
Call the broadest range of structural variant types using whole genome sequencing with high precision and recall.
SMRT Analysis
Structural variant calling is a streamlined application available through the SMRT Link graphical user interface and command line. Use this application to call variants and visualize results with the integrative genomics viewer (IGV).
How it works
The reads are mapped to the reference sequence then variants are called.

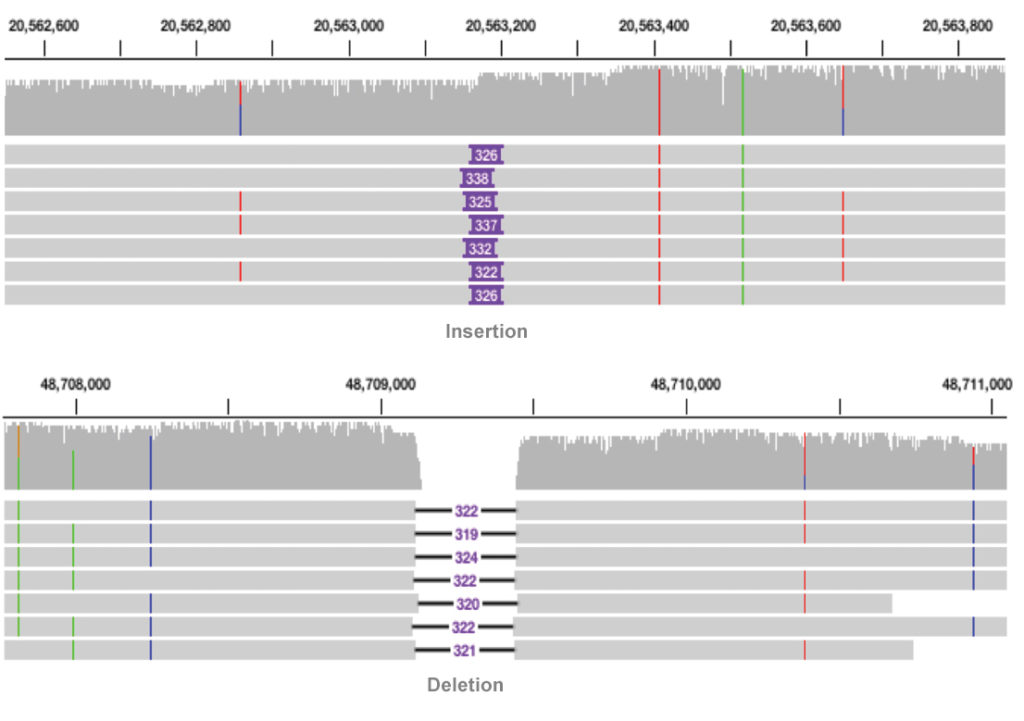
Learn more about SMRT Analysis and structural variant calling
PacBio DevNet tools
There are several community-supported tools for structural variant calling using SMRT sequencing data. They provide scientists with easy access to additional open-source algorithms such as:
Sniffles
Calls all types of structural variants using evidence from split-read alignments, high-mismatch regions, and coverage analysis
SMRT-SV
Calls insertions, deletions, and inversions using a local assembly approach
SMRT compatible analysis products
Our analysis partners offer several solutions for variant calling. Options include customized analysis based on project needs, full-service genomics analytics, cloud-based platforms with open source tools, or opportunity to run your own algorithms.
DNAnexus
Cloud-based genome informatics platform that provides data analysis tools and structural variant calling using pbsv, PBHoney, Sniffles, and the Parliament suite
Computomics
Custom genome analysis services including structural variant calling
See our complete list of SMRT compatible analysis products