The recent beta release of version 3 of the popular genome browser IGV greatly improves support for PacBio data [1]. The long reads (up to 50 kb) and random error profile of PacBio SMRT® sequencing facilitate new applications in genome assembly, structural variant discovery, and haplotype phasing. These unique properties and applications benefit from customized data visualization.
IGV 3 extends support for PacBio long reads with: performance improvements to enable viewing variants at multi-kilobase scales; a “quick consensus” mode that suppresses single read random errors; labels for large insertion and deletion structural variants; and “group by base” to explore haplotype phase. The new capabilities are featured in a 4-minute tutorial video. To try them yourself, upgrade to Java 8, install IGV 3, and then load this IGV session (File > Open Session) with a sample dataset of 70-fold sequencing of a human genome, HG002 from NIST Genome in a Bottle [2].
Quick Consensus
It is visually challenging to identify biological variation (single nucleotide variants occur about every 1,000 basepairs in humans) among the more frequent sequencing errors in PacBio reads. However, because PacBio errors are random, quality is extremely high in a consensus of independent reads, often surpassing the quality from next-generation sequencing [3]. A mismatch that is consistent across reads indicates biological variation [4].
IGV has added two features, “quick consensus mode” and “hide indels”, to reveal biological variation in PacBio reads. The quick consensus mode shows mismatches only at positions where more than a specified fraction of reads disagrees with the reference (recommended setting: 25%). The logic is the same as used by the coverage track. The “hide indels” feature (recommended setting: <10 bp) suppresses the most common error in raw PacBio reads, random small indels. Both features are available in the “Alignment” tab of the IGV preferences (View menu > Preferences).
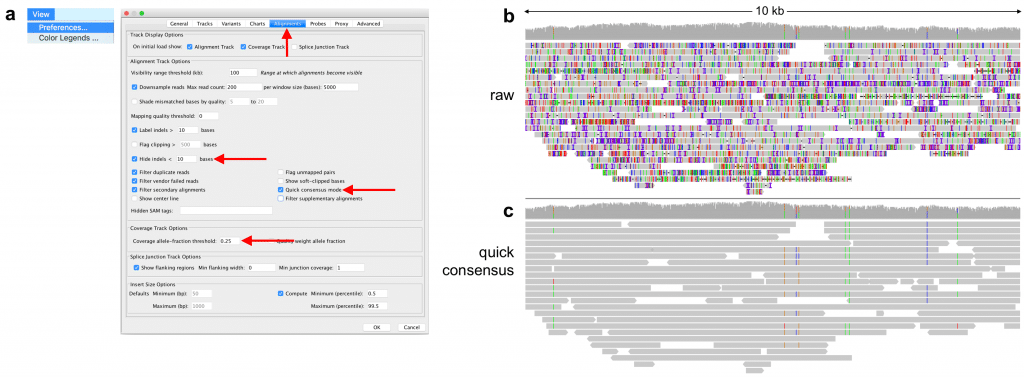
Figure 1. Quick consensus mode and indel hiding reveal biological variation in PacBio long reads. (a) Both quick consensus and indel hiding are available in the “Alignment” tab of the IGV preferences (View menu > Preferences). Recommended settings are to hide mismatches at below 25% coverage allele fraction and indels shorter than 10 bases. (b) Raw PacBio reads with no consensus error correction. (c) The same read data with consensus mode and indel hiding activated reveals a number of homozygous and heterozygous single nucleotide variants.
Structural Variants
Each human genome has approximately 20,000 structural variants (differences ≥50 basepairs with the reference), most of which require PacBio long reads to detect [5]. For variants contained in a single read alignment, IGV 3 adds an option to “label large indels”, which lists the basepair size of the variant on a colored block whose width is proportional to the size of the indel. The “label large indels” feature is available in the “Alignment” tab of the preferences (View menu > Preferences). The recommended setting is to label indels larger than 10 basepairs.
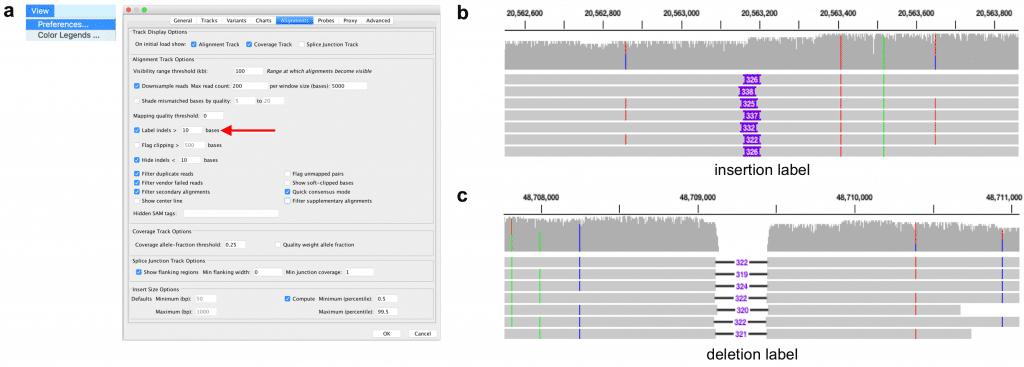
Figure 2. Label insertion and deletion structural variants. (a) The option to label large indels is available in the “Alignment” tab of the IGV preferences (View menu > Preferences). Recommended settings are to label indels larger than 10 bases. (b) An insertion larger than the defined threshold is indicated by a purple box. The width of the box is proportional to the size of the insertion, and the basepair size is written on the box if it fits. (c) A deletion is indicated by a black line. The basepair size of the deletion is written on a white box at the center of the line. Examples are from HG002 sequenced by Genome in a Bottle.
For reads with very large structural variants or which contain inversions, mappers like BWA produce separate primary and supplementary alignments. IGV 3 adds an option to “link supplementary alignments” to visually connect separate alignments from the same read. For reads that align to both strands, which can indicate an inversion, forward alignments are colored pink and reverse alignments are colored blue. “Link supplementary alignments” is available in the right-click menu for each alignment track.
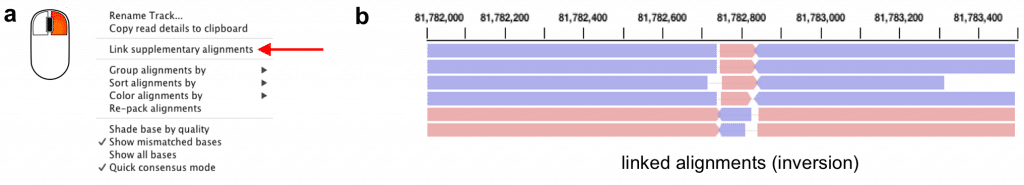
Figure 3. Link alignments from the same read. (a) The option to link primary and supplementary alignments from a read is available in the right click menu for the alignment track. (b) When “link alignments” is active, separate alignments from the same read are drawn on a single row and connected by a thin line. For reads that align to both strands, which can indicate an inversion, forward alignments are colored pink and reverse alignments are colored blue. The example shows reads that support an inversion in HG002.
Haplotype Phasing
PacBio long reads can span multiple single nucleotide and structural variants, which directly phases the variants into haplotypes [6]. To support visual exploration of haplotypes, IGV 3 adds an option to “group by base,” which categorizes reads by the basepair at a selected position. “Group by base” is available by right clicking on the basepair position by which to group. IGV 3 also includes performance improvements that enable variation to be shown at zoom levels of 10 kb and larger, which is critical to view haplotype structure.
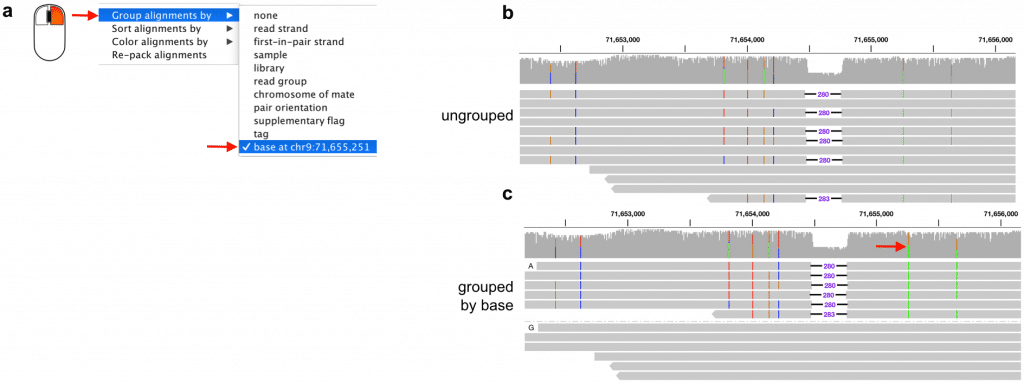
Figure 4. Explore haplotype phase by grouping alignments by basepair. (a) The option to group alignments by the basepair at a selected position is available in the right click menu for the alignment track. (b) Ungrouped alignments from a locus in HG002 with a heterozygous deletion and several heterozygous single nucleotide variants. (c) Grouping the alignments by a heterozygous single nucleotide variant reveals two clear haplotypes.
To utilize the new capabilities, upgrade to Java 8, install IGV 3, and then load this IGV session (File > Open Session) with a sample dataset of 70-fold sequencing of a human genome, HG002. Congratulations to Jim Robinson, Helga Thorvaldsdóttir, and the rest of the IGV team and community for the release of IGV 3!
[1] Robinson JT, et al. (2011). Nat Biotechnol, 29(1):24-6.
[2] Zook JM, et al. (2016). Sci Data, 3:160025
[3] Roberts RJ, et al. (2013). Genome Biol, 14(7):405.
[4] Chin CS, et al. (2013). Nat Methods, 10(6):563-9.
[5] Chaisson MJ, et al. (2016). Nature, 517(7536):608-11.
[6] Chin CS, et al. (2016). Nat Methods, 13(12):1050-4.