
A map of every individual’s genome will soon be possible, but how will we know if it is correct? Benchmarks are needed in order to check the performance of sequencing, and any genomes used for such a purpose should be comprehensive and well characterized.
Enter the Genome in a Bottle Project (GIAB), a consortium of geneticists and bioinformaticians committed to the creation and sharing of high-quality reference genomes. Unlike other initiatives, such as the 1000 Genomes Project, that are seeking to sequence many representatives of different populations, GIAB is interested in sequencing just a few individuals, but deeply and with multiple technologies. Formed in 2012, the consortium has to date released data for five individuals, including an Ashkenazi Jewish family trio.
GIAB has made great progress with characterizing small variants, such as SNPs and indels. However, as project co-leader Justin Zook explained in a presentation at the Labroots Genetics and Genomics conference, much work remains to be done. Zook estimates that the current GIAB truth sets, based mostly on short-read sequencing, miss 200,000 variants in tandem repeats and homopolymers. Further, the vast majority of medium and large variants are missed: over 75% for indels 15-50 bp and 99% for structural variants >50 bp.
“The representation of these variants is poorly standardized, and that’s especially true once you get to more complex changes that occur in repetitive regions,” Zook said. “And tools to do the comparisons for structural variants are really in their infancy.”
The solution? New technologies like accurate, long reads from SMRT Sequencing, and new variant callers, especially those based on de novo assembly. Zook, a scientist at the National Institute of Standards and Technology, and the GIAB consortium are currently applying these techniques to build benchmark sets of structural variants. Using PacBio long reads, the GIAB consortium has expanded its structural variant callset from only a few hundred variants to over 20,000.
“When we’re trying to characterize the structural variation in long, repetitive regions, or in places where there are large insertions, it’s been really useful to have long-read information,” Zook said. “Long reads are also really useful for phasing variants, and it looks like they’ll be really useful for characterizing variants in difficult-to-map regions,” he added.
In addition to providing completed benchmark reference genomes, GIAB also releases datasets; a 2016 release included 12 datasets based on seven genomes, compiled by 51 authors from 14 institutions.
Zook said new public long-read datasets are coming. The data in development includes SMRT Sequencing of a Chinese trio (in collaboration with the Icahn School of Medicine), and a deeper dive into the genomes of the Ashkenazi Jewish son and mother of the originally released trio set.
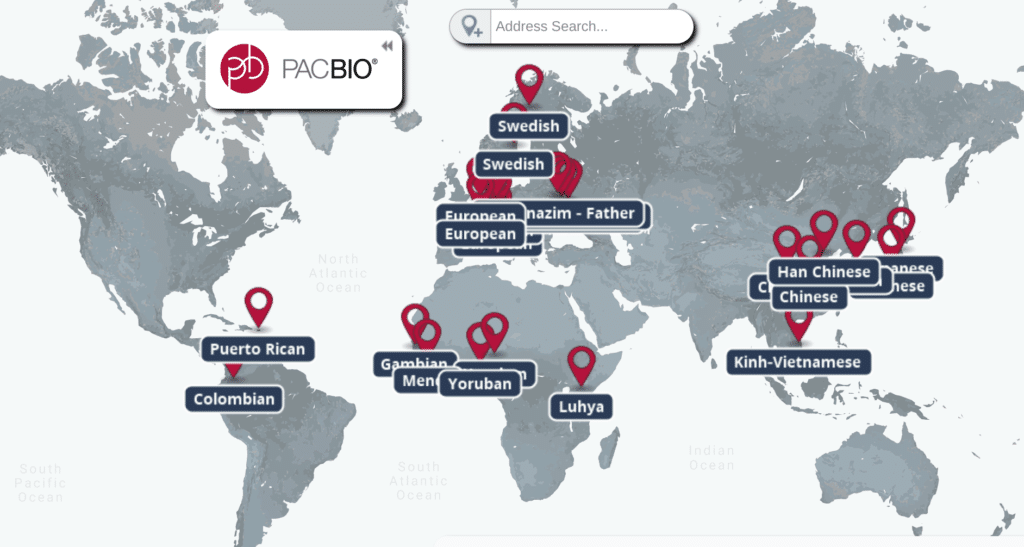
You can watch Zook’s complete presentation on the Labroots site. Also, check out our interactive map to learn more about the many population-specific human genomes generated by SMRT Sequencing.
September 4, 2018 | Events + conferences